pLM-SAV: A Δ-Embedding Approach for Predicting Pathogenic Single Amino Acid Variants
pLM-SAV: A Δ-Embedding Approach for Predicting Pathogenic Single Amino Acid Variants
Gereben, O.; Tordai, H.; Khamisi, L.; Hegedus, T.
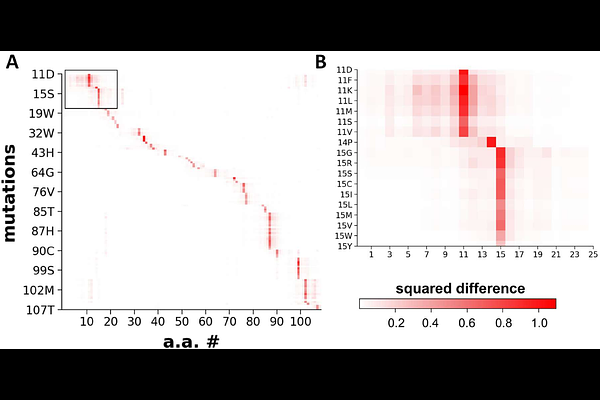
AbstractPredicting whether single amino acid variants (SAVs) in proteins lead to pathogenic outcomes is a critical challenge in molecular biology and precision medicine. Experimental determination of the effects of all possible mutations or those observed in pathogenic individuals is infeasible. While existing state-of-the-art tools such as AlphaMissense show promise, their performance remains insufficient for diagnostic applications, they are often challenging to run locally. To address these limitations, we developed pLM-SAV, a simple yet effective predictor leveraging protein language models (pLMs). Our method computes delta-embeddings by subtracting the embedding of the mutant sequence from that of the wild type sequence. These delta-embedding vectors serve as input for a convolutional neural network used for training and prediction. To prevent data leakage, we trained our model on a well-characterized, labeled set of Eff10k and evaluated it on a non-homologous subset of ClinVar data. Our results demonstrate that this approach performs exceptionally well on the Eff10k test folds and reasonably on ClinVar test sets. Notably, pLM-SAV excels in resolving ambiguous predictions by AlphaMissense. We also found that an ensemble method, REVEL, outperforms both AlphaMissense and pLM-SAV, thus, we integrated these REVEL-enhanced predictions into our widely used AlphaMissense web application, https://alphamissens.hegelab.org. Our results demonstrate that an SAV predictor trained on labeled data can achieve high predictive performance. We anticipate that incorporating delta-embeddings into other mutation effect predictors or mutant structure prediction methods will further enhance their accuracy and utility in diverse biological contexts.