ViralMap: Predicting Features in Viral Proteins from Primary Sequence
ViralMap: Predicting Features in Viral Proteins from Primary Sequence
Dwivedi, S.; Kar, S.; Horton, A. P.; Gollihar, J. D.
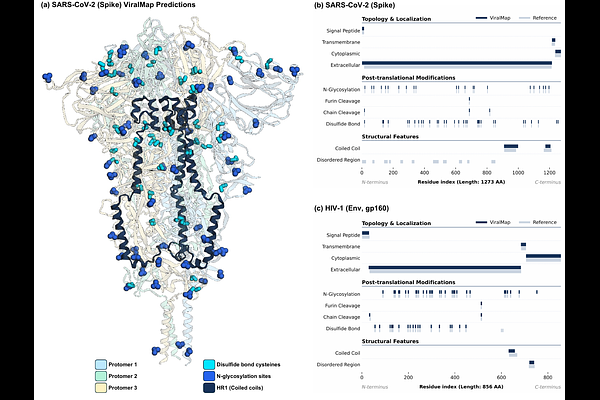
AbstractModern viral vaccines are designed to elicit an immune response against viral proteins that mediate infection, making those proteins important targets for characterization and engineering. To improve vaccine efficacy, the proteins often require changes to specific residues or domains to improve immunogenicity and induce a protective response. These engineering strategies vary significantly across viruses, and comprehensive and accurate protein sequence annotation is crucial for guiding vaccine design. The growing risk of novel pathogen emergence and initiatives such as the CEPI 100 Days Mission to rapidly counter "Disease X" threats heighten the need for tools that can convert viral protein sequences from newly characterized genomes or emerging variants into the annotation profiles required for antigen engineering. To address this, we developed ViralMap, a multi-label annotation model tailored for eukaryotic viral proteins. By leveraging ESM-2 language model representations, ViralMap simultaneously predicts ten distinct annotation classes spanning domain topology and localization, post-translational modifications, and structural features directly from primary sequences. The model achieves a residue-level precision-recall area under the curve (PR-AUC) of 0.75 or greater for seven of the ten classes and realizes predictive performance competitive with established tools across the eight benchmarked classes. Evaluation on complex glycoproteins, including the SARS-CoV-2 spike and HIV-1 Env, supported cross-strain and novel-family generalization. By providing a unified, sequence-based framework for multi-label annotation, ViralMap offers a practical and scalable bridge from raw viral protein sequences to the annotation profiles required for antigen engineering.