Antimuscarinic drugs exert β-arrestin-biased agonism at the muscarinic acetylcholine type 1 receptor
Antimuscarinic drugs exert β-arrestin-biased agonism at the muscarinic acetylcholine type 1 receptor
Amiri, S.; Aghanoori, M.-R.; Smith, D. R.; Waise, T. M. Z.; Lao, Y.; Inoue, A.; Zahedi, R.; Dunn, H. A.; Fernyhough, P.
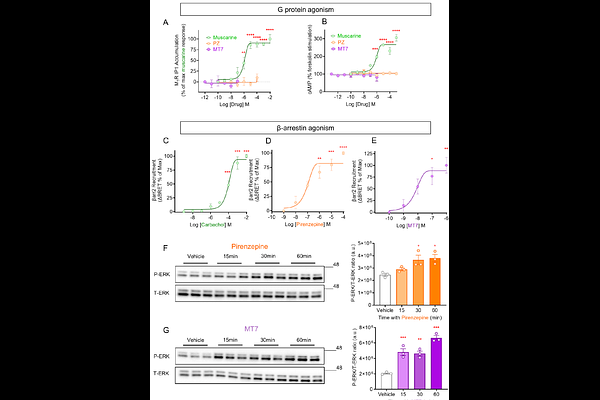
AbstractPrevious studies indicate that both pirenzepine (PZ), a selective orthosteric muscarinic acetylcholine type 1 receptor (M1R) antagonist, and muscarinic toxin 7 (MT7), a negative M1R allosteric modulator (NAM), act via M1R to promote neuritogenesis in cultured adult rodent primary dorsal root ganglia (DRG) sensory neurons, in part, through beta-arrestin-dependent activation of extracellular signal-regulated protein kinase 1/2 (ERK1/2). Furthermore, these antagonists reverse nerve degeneration in a variety of rodent models of peripheral neuropathy through multiple complementary pathways. To understand the therapeutic effects and mechanism of M1R antagonist-induced ERK1/2 phosphorylation, we tested the hypothesis that PZ and MT7 possess beta-arrestin-biased agonism at M1R to drive activation of ERK and enhance neurite outgrowth. Treatment for up to 30 min with PZ and MT7 dose-dependently recruited beta-arrestin2 to M1R (analyzed using nano-BRET) and increased ERK phosphorylation in both HEK293 cells and DRG neurons. DRG neurons of different sub-types express M1R, and ERK activation by MT7 was only observed in M1R-positive neurons. These novel pharmacological effects occurred in the absence of activation of G protein signaling or receptor internalization. PZ phosphorylated M1R at six specific serine/threonine residues (T230, S251, T254, S321, T354, S356) of intracellular loop 3 (ICL3) and deletion mutation of these sites suppressed PZ and MT7 induction of beta-arrestin binding to M1R and inhibited ERK activation. With regard to PZ signaling, alanine substitution at S251 and T254 was sufficient to impede beta-arrestin binding and ERK activation. Beta-arrestin-biased activity of PZ and MT7 involved the mobilization of casein kinase 2 (CK2) and this occurred in the absence of Galphaq or G protein receptor kinase (GRK) activity. Pharmacological or siRNA-based inhibition of CK2 blocked PZ-induction of beta-arrestin association, ERK activation and neurite outgrowth in DRG neurons. In conclusion, PZ/MT7 activated M1R toward the beta-arrestin signaling pathway in both HEK293 cells and DRG neurons to augment ERK activation and neurite outgrowth via engagement of CK2.