Proteome-wide multi-omics profiling of osteosarcoma transcription factor networks
Proteome-wide multi-omics profiling of osteosarcoma transcription factor networks
Thang, N. X.; Martiensen, E. L. B.; Abdelhalim, M.; Tran, T. T.; Ledsaak, M.; Rogne, M.; Thiede, B.; Eskeland, R.
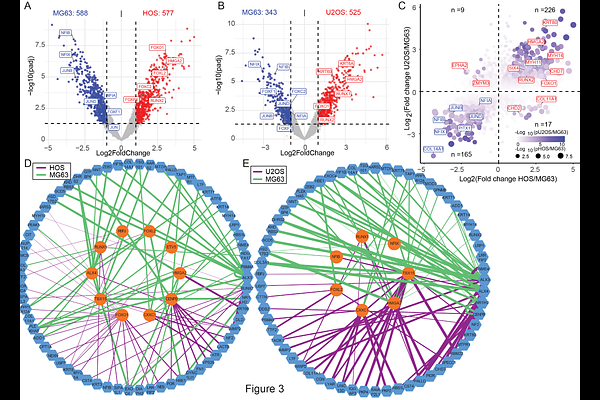
AbstractOsteosarcoma (OS) is an aggressive bone cancer that most commonly affects children and young adults. OS exhibits a high degree of genomic complexity, as well as cellular plasticity, and dynamic transcriptional regulation is suggested to contribute to treatment resistance and metastasis. Cell lines are well characterized as models to advance our knowledge on OS biology. HOS and U2OS cells have increased invasiveness and higher migratory ability compared with MG63. In this study, we employed a tandem array of consensus transcription factor response elements (catTFREs) proteomic approach to characterize transcription factor (TF) regulatory networks related to OS aggressiveness. We mapped 7,594 proteins and enriched 352 transcription factors and coregulators. When we integrated proteomics with cell line specific gene expression and chromatin accessibility we classified the proteins into different OS cell line dependent sub-clusters and identified TFs and coregulators common for all cell lines and specific for individual cell lines. We demonstrate that RUNX2, MYBL2 and HMGA2 are specifically enriched in HOS and U2OS and may be linked to the cell aggressiveness. ETV5, JUNB, NFIX and ZEB1 were among TFs specific to MG63. Our analysis provides a more comprehensive understanding of the transcriptional drivers that shape OS regulatory landscapes and may have future therapeutic implications.