A Chk1-Sp1-CD59 axis of the DNA damage response impedes rituximab-mediated complement-dependent cytotoxicity
A Chk1-Sp1-CD59 axis of the DNA damage response impedes rituximab-mediated complement-dependent cytotoxicity
Chan, A. S. Y.; Jaynes, P. W.; Anbuselvan, A.; Ong, C. Z. Y.; Hoppe, M. M.; Yong, W. K.; Khanchandani, V.; Lee, J. M.; Mustafa, N.; Azaman, I.; Hoang, P. M.; Hong, G.; Chng, W. J.; Cragg, M. S.; Kappei, D.; Tripodo, C.; Jeyasekharan, A. D.
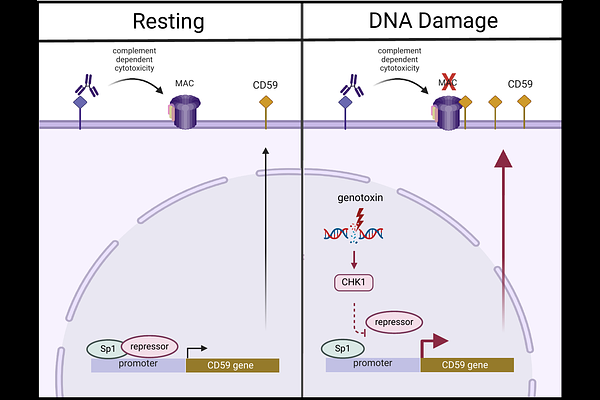
AbstractDNA damage responses primarily coordinate intracellular processes, but their effects on the cell surface remain poorly understood. We uncovered a DNA damage response at the cell surface that regulates complement dependent cytotoxicity (CDC), a key mechanism of antibody-mediated tumor cell killing. We found that several mCRPs, such as CD46, CD55, and CD59, are upregulated in diffuse large B-cell lymphoma (DLBCL) cells following chemotherapy. These mCRPs protect cells from CDC, including CDC triggered by anti-CD20 immunotherapy. Chemotherapy pre-treatment reduced the efficacy of rituximab-induced CDC, with CD59 exerting a dominant function. A high-throughput kinase screen revealed that Chk1 inhibition restored CDC sensitivity. Mechanistically, chemotherapy-induced CD59 expression was Chk1-dependent and regulated at an Sp1-driven promoter site. Inhibition of Sp1 with Mithramycin A abrogated chemotherapy-induced CD59 transcription. Co-immunoprecipitation mass spectrometry revealed that DNA damage releases transcriptional repressors from Sp1, an effect reversed by Chk1 inhibition. These findings define a Chk1-Sp1-CD59 signaling axis linking genotoxic stress to complement modulation. Our study reveals potential antagonism between chemotherapy and antibody-mediated CDC and highlights consideration in sequencing combination therapies and a therapeutic potential of Chk1 inhibitors in maintaining CDC efficacy during chemoimmunotherapy.