RIP2 inhibits DOCK8-mediated platelet dense granule release and alleviates experimental myocardial infarction
RIP2 inhibits DOCK8-mediated platelet dense granule release and alleviates experimental myocardial infarction
Zhang, J.; Zhang, Z.; Pan, G.; Liu, Y.; Chang, L.; Qi, Z.; Zhang, Y.; Zhang, S.; Qiao, J.; Xi, X.; Dai, K.; Dong, J.; Ding, Z.
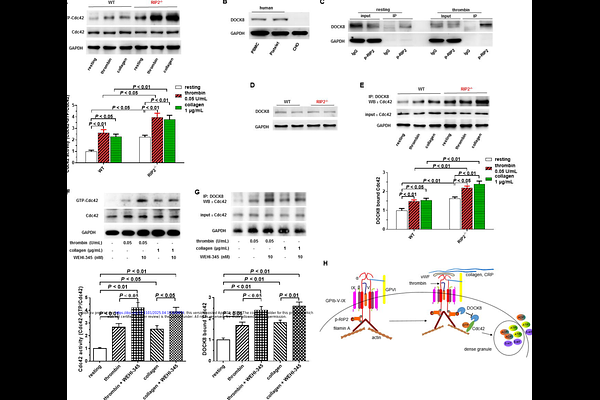
AbstractObjective: Receptor-interacting protein 2 (RIP2) is an essential mediator of inflammation and innate immunity downstream of pattern recognition receptors (PRRs). Platelets express RIP2, while its role in platelet activation, thrombosis, and myocardial infarction (MI) is unknown. Approach and Results: Here we show that RIP2 deficiency enhances platelet dense granule secretion in response to GPIb and GPVI activation; platelet aggregation in whole blood and adhesion under arterial shear are also increased. Consistently, RIP2 inhibitor WEHI-345 potentiates human platelet dense granule secretion and inhibits RIP2 phosphorylation induced by thrombin and collagen. These phenotypes are translated into shorter bleeding time, accelerated FeCl3-induced arterial thrombosis. Importantly, platelets from patients with coronary artery disease and mice with atherosclerosis express lower RIP2, and RIP2 deficiency deteriorates MI and cardiac function in a mouse ischemia/reperfusion model. Mechanistically, we found that platelets express dedicator of cytogenesis protein 8 (DOCK8), which is sequestered by phosphorylated RIP2 (p-RIP2), causing inhibition of Cdc42 activation and subsequent dense granule release. Conclusions: In conclusion, RIP2 inhibits platelet activation, thrombosis, and ameliorates MI. Our results also suggest that a novel PRR-independent pathway, p-RIP2-DOCK8-Cdc42, negatively regulates platelet activation downstream of GPIb and GPVI. Platelet RIP2 pathway may be promising for therapeutic intervention of atherothrombotic diseases from early atherosclerosis stage to arterial thrombosis and MI.