Guide-tree bias of whole genome alignment can mislead phylogenomic analyses
Guide-tree bias of whole genome alignment can mislead phylogenomic analyses
Tao, Q.; Grünewald, S.
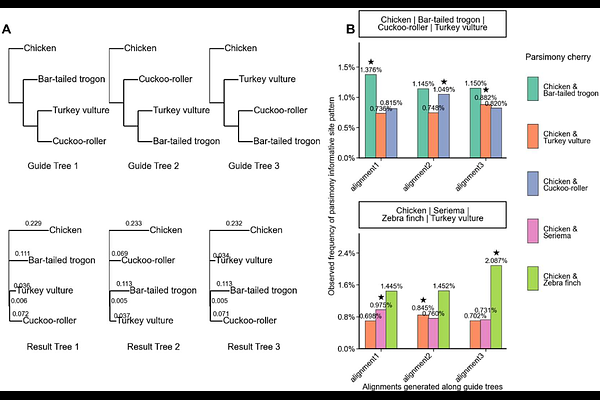
AbstractWhole-genome alignment (WGA) is widely used for genome-scale phylogenetic inference, and most scalable WGA pipelines rely on progressive alignment guided by a pre-specified tree. Among progressive whole-genome aligners, Progressive Cactus is a successful state-of-the-art method. However, analyses of real and simulated avian data indicate that guide-tree choice can influence downstream tree inference; star guide trees do not remove this effect and can exacerbate long-branch attraction artefacts. We have developed a consensus strategy based on the Progressive Cactus framework by generating a small set of alternative guide-tree alignments and retaining only homology relationships consistently recovered across all alignments. In simulation experiments, consensus alignments improve precision, bring inferred site-pattern frequency distributions closer to those of the true alignments, and recover more true splits than single guide-tree alignments. In a real landbird (Telluraves) dataset, we observe a strong bias towards single binary guide trees and long-branch attraction for less resolved trees. While the reconstructed tree still depends on the phylogenetic method and taxa sampling, our consensus alignment has no clear bias. We implemented a hierarchical consensus workflow that only locally resolves uncertainty in the guide tree. Therefore, the computational cost increases only moderately, for example by an estimated 68 percent for a recently published large-scale alignment of more than 300 modern birds (Neoaves) taxa.