Hi-Compass resolves cell-type chromatin interactions by single-cell and spatial ATAC-seq data across biological scales
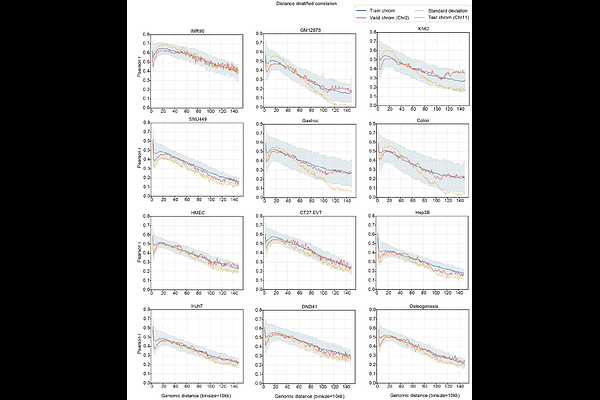
Hi-Compass resolves cell-type chromatin interactions by single-cell and spatial ATAC-seq data across biological scales
Sun, Y.-C.; Jiang, W.-J.; Cai, K.-W.; Wu, H.-J.
AbstractComputational prediction of three-dimensional (3D) genome organization provides an alternative approach to overcome the cost and technical limitations of Hi-C experiments. However, current Hi-C prediction models are constrained by their narrow applicability to studying the impact of genetic variation on genome folding in specific cell lines, significantly restricting their biological utility. We present Hi-Compass, a generalizable deep learning model that accurately predicts chromatin organization across diverse biological contexts, from bulk to single-cell samples. Hi-Compass outperforms existing methods in prediction accuracy and is generalizable to unseen cell types through chromatin accessibility data, enabling broad applications in single cell omics. Hi-Compass successfully resolves cell-type-specific 3D genome architectures in complex biological scenarios, including immune cell states, organ heterogeneity, and tissue spatial organization. Furthermore, Hi-Compass enables integrative analysis of single-cell multiome data, linking chromatin interaction dynamics to gene expression changes across cell clusters, and mapping disease variants to pathogenic genes. Hi-Compass also extends to spatial multi-omics data, generating spatially resolved Hi-C maps that reveal domain-specific chromatin interactions linked to spatial gene expression patterns.