A scalable computational framework for annotation-independent combinatorial target identification in scRNA-seq databases
A scalable computational framework for annotation-independent combinatorial target identification in scRNA-seq databases
Buser, M.; Marr, C.
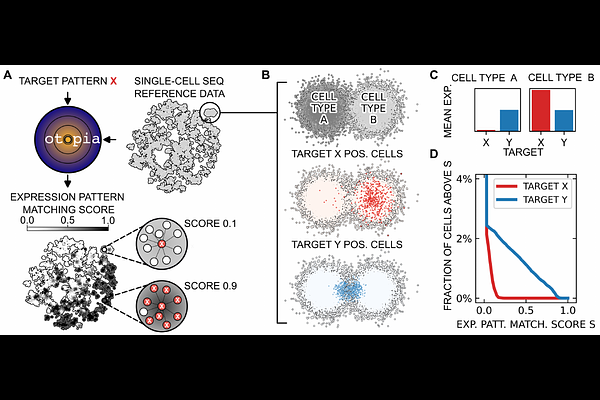
AbstractThe applicability of novel targeted cell therapies is constrained by on-target off-tumor toxicities, posing significant challenges for safe and effective treatment. To develop strategies for mitigating these toxicities, a comprehensive understanding of the distribution of single-cell profiles across human tissues is essential. Single-cell RNA sequencing (scRNA-seq) provides detailed insights into cellular diversity and function. However, as data collection expands and RNA expression atlases comprise tens of millions of cells, effectively querying these databases to identify cell populations matching specific gene expression patterns becomes challenging. Key challenges include managing data volume, handling batch effects, navigating study-dependent data curation and annotation, and addressing the combinatorial complexity of possible target patterns. User-friendly screening tools that allow for complex query target patterns without relying on existing annotations are currently missing. Here we present otopia, a computational framework that enables fast queries of large scRNA-seq databases, enhancing the precision of cell population identification. Using precomputed neighborhood graphs and boolean logic for expression patterns, otopia enables efficient queries addressing arbitrary combinatorial target patterns. otopia does not depend on cell type annotations and by exploiting unbiased expression pattern matching scores, it uncovers critical populations that annotation based methods overlook. We exemplify otopia\'s capabilities by identifying the largest B cell populations in the CELLxGENE census data using a CD19+CD22+CD5- query target pattern. Moreover, we show that otopia can provide a concise overview of cell populations matching hypothetical acute myeloid leukemia (AML) dual targets, realizing logic AND gates, across different tissues. otopia enables the estimation of on-target off-tumor toxicities for drugs targeting both single markers and complex marker combinations, surpassing previous methods that focus mainly on single markers. With the expanding availability of scRNA-seq data, otopia represents a scalable, unbiased, and automated approach to refine putative therapeutic targets in order to minimize toxicities.