Chemotherapy induces tissue NAD+ loss, and downregulation of NAD+ biosynthetic enzyme Nrk2 marks muscle wasting
Chemotherapy induces tissue NAD+ loss, and downregulation of NAD+ biosynthetic enzyme Nrk2 marks muscle wasting
Poellaenen, N.; Gammon, C.; Pin, F.; Huot, J.; Sartori, R.; Penna, F.; Hulmi, J. J.; Bonetto, A.; Pirinen, E.
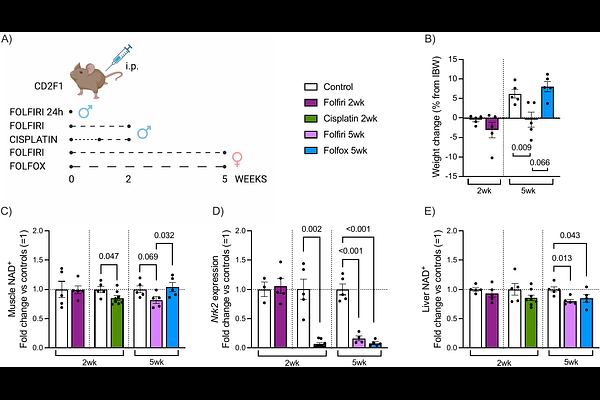
AbstractBackground Aberrant NAD+ metabolism has been implicated in the pathogenesis of cancer cachexia, highlighting this pathway as a potential therapeutic target to mitigate skeletal muscle wasting. However, it remains unclear whether chemotherapeutic agents contribute to the onset of cachexia by disrupting NAD+ metabolism. Here, we investigated the effects of commonly used chemotherapy regimens on NAD+ metabolism in skeletal muscle and liver of healthy mice. Methods Healthy mice were treated with either 2-week regimens of folfiri or cisplatin, or 5-week regimens of folfiri or folfox, with vehicle-treated mice serving as controls. Cachexia-related outcomes were assessed, while skeletal muscle and liver tissues were analyzed for NAD metabolites and markers of NAD+ metabolism. Given the consistent downregulation of the NAD+ biosynthetic enzyme Nrk2 in cachectic chemotherapy-treated mice, we examined skeletal muscle Nrk2/NRK2 expression across published murine and human cachexia datasets, and in additional models of muscle wasting and hypertrophy. Results NAD+ loss was observed in atrophic muscle following administration of cisplatin (2-week treatment; -14% vs controls, p=0.047) and folfiri (5-week treatment; -18%, p=0.069). In contrast, muscle NAD+ levels were preserved in non-atrophic groups (2-week folfiri and 5-week folfox). Muscle Nrk2 was the most responsive NAD+ biosynthetic enzyme, showing consistent downregulation across chemotherapy models with ongoing or developing muscle loss: cisplatin (-93%, p<0.001), folfiri (-84%, p<0.001) and folfox (-92%, p<0.001). In the liver, NAD+ levels declined after prolonged 5-week folfiri (-20%, p=0.013) and folfox (-15%, p=0.043) treatments. These changes were accompanied by distinct alterations in NAD+ biosynthesis pathways, indicating treatment-specific reorganization of hepatic NAD+ metabolism. Cross-study analyses revealed early and consistent skeletal muscle Nrk2 downregulation across multiple murine cachexia models and human inactivity studies, whereas cachexia-targeted interventions in rodents and resistance training in humans increased its expression. Conclusions These findings demonstrate that chemotherapy disrupts tissue NAD+ metabolism, with skeletal muscle NAD+ loss accompanying muscle atrophy and hepatic NAD+ levels declining after prolonged treatment. The early and robust responsiveness of muscle Nrk2 expression to changes in muscle mass underscores its potential as a dynamic indicator for predicting treatment-induced changes in muscle mass. Together, these results provide new molecular insight into the metabolic basis of chemotherapy-induced muscle wasting and support further investigation of NAD+-targeted strategies in this context.