Deconvolution of multiplexed peptidoform mass spectra enables high-resolution profiling of complex protein modification patterns
Deconvolution of multiplexed peptidoform mass spectra enables high-resolution profiling of complex protein modification patterns
Cheng, Z.; Zhai, L.; Yu, L.; Chen, K.; Zhao, W.; Tan, M.; Fu, Y.
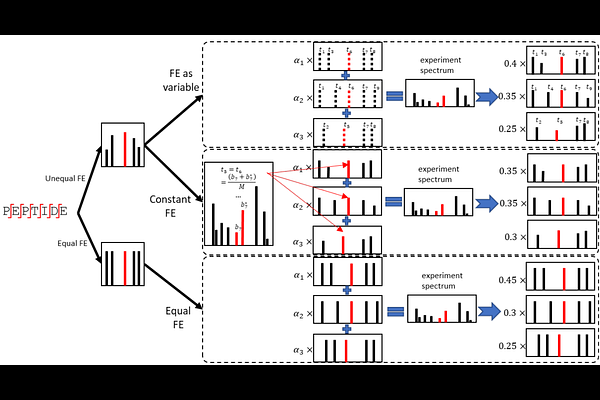
AbstractCombinatorial patterns of post-translational modifications (PTMs), like the histone code, play critical roles in regulating protein functionality. However, in bottom-up proteomics, isobaric modification peptidoforms (IMPs) frequently co-elute and co-fragment during liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS), resulting in multiplexed mass spectra that cannot be properly analyzed by conventional protein identification and quantification methods. Existing solutions are either limited to highly narrow scenarios or unavailable for public use. Here, we introduce PTMdecoder, a robust and versatile software tool for discriminating and quantifying IMPs by deconvoluting their multiplexed MS/MS spectra and ion chromatograms without requiring a spectral library of individual peptidoforms. Tested on 34 samples of synthesized IMPs, PTMdecoder achieved an overall sensitivity of 95.7% for IMP identification and root mean square errors mostly <0.05 for quantification. In human histone samples, it detected and quantified 36.7-44.8% more IMPs than conventional methods, providing a higher-resolution view of histone modification patterns.