A method for massively scalable phylogenetic network inference
A method for massively scalable phylogenetic network inference
Kolbow, N.; Kong, S.; Solis-Lemus, C.
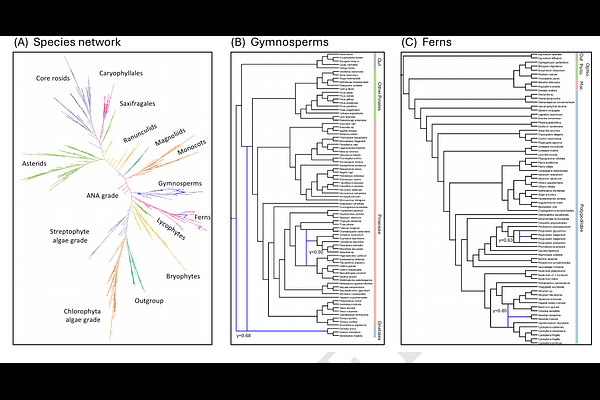
AbstractRecent advancements in sequencing technologies have enabled large-scale phylogenomic analyses. While these analyses often rely on phylogenetic trees, increasing evidence suggests that non-treelike evolutionary events, such as hybridization and horizontal gene transfer, are prevalent in the evolutionary histories of many species, and in such cases, tree-based models are insufficient. Phylogenetic networks can capture such complex evolutionary histories, but current methods for accurately inferring them lack scalability. For instance, state-of-the-art model-based approaches are limited to around 30 taxa. Implicit network inference methods like NeighborNet and Consensus Networks are fast but lack biological interpretability. Here, we introduce a novel method called InPhyNet that merges a set of non-overlapping, independently inferred networks into a unified topology, achieving linear scalability while maintaining high accuracy under the multispecies network coalescent model. Our simulations show that InPhyNet matches the accuracy of SNaQ on datasets with 30 taxa while drastically decreasing the overall network inference time. InPhyNet is also more accurate than implicit network methods on large datasets while maintaining computational feasibility. Re-analyzing a phylogeny of 1,158 land plants with InPhyNet, we recover known reticulate events and provide evidence for the controversial placement of Order Gnetales within gymnosperms. These results demonstrate that InPhyNet enables biologically meaningful network inference at previously unprecedented scales.