DNA methylation networks during pig fetal development: a joint fused ridge estimation approach
DNA methylation networks during pig fetal development: a joint fused ridge estimation approach
Wachala, K. M.; de Vos, J.; Madsen, O.; Derks, M. F. L.; Peeters, C. F. W.
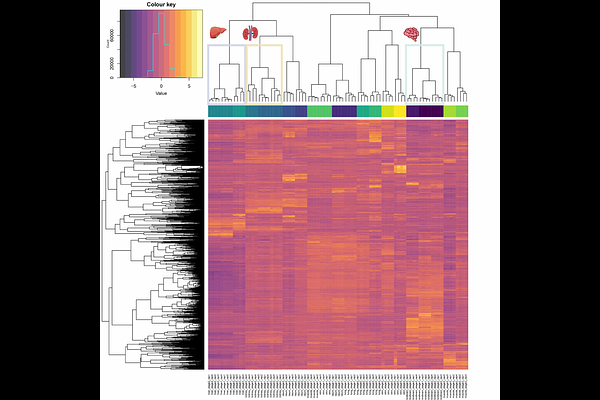
AbstractAlthough an organisms genetic information is predominantly identical among most of its cell types, the epigenome regulates the expression of the genome in a cell type- and context-dependent manner. In mammals, DNA methylation in regulatory regions, such as promoters, primarily regulates gene expression by inducing transcriptional inactivation. With genome-wide approaches came the realization that DNA methylation patterns underlying mammalian development are considerably more dynamic than previously recognized. This realization highlights the need for methodological approaches capable of capturing this phenomenon. In this study, we investigated the feasibility of modeling DNA methylation networks by jointly estimating regularized precision matrices from time- and tissue-specific omics data derived from the pig genome. For that, we utilized RNA-Seq and RRBS data that span seven pig tissues at three developmental stages: early organogenesis, late organogenesis, and newborn. Our analysis focused on 61, 48, and 74 genes -- differentially expressed across developmental stages and CpG-methylated in promoter regions, from endoderm-, mesoderm-, and ectoderm-derived tissues, respectively. Using a joint fused ridge approach, we were able to borrow information across tissues and time points, enabling more robust network inference. This analytical framework advances exploratory methods for studying organism development using pig as a model species. Our results highlight the importance of fetal-maternal immunity and the circulatory system in early development, and shed light on dynamic interactions across tissues, organ systems, and germ layers. We anticipate that this flexible framework can be extended to other omics data and species, facilitating future research.