Three phylogenetic metrics are compatible with natural evolution of the earliest SARS-CoV-2 sequence
Three phylogenetic metrics are compatible with natural evolution of the earliest SARS-CoV-2 sequence
Lorenzi, J.-N.; Graner, F.; Bigot, T.; Decroly, E.; Courtier-Orgogozo, V.; Achaz, G.
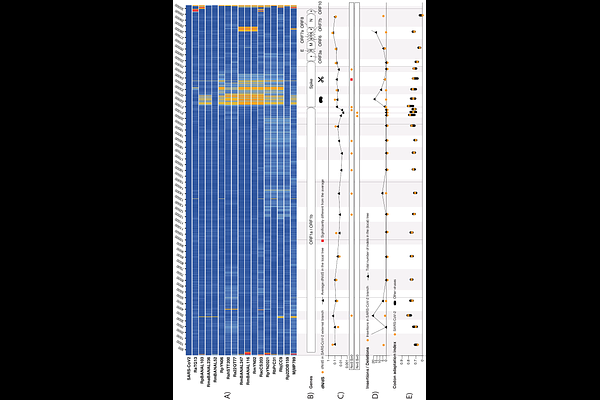
AbstractComparative analyses of coronavirus sequences can unravel important aspects of their complex evolution. Here we develop a method to infer past recombination breakpoints based on minimizing homoplasies count. We test it on three outbreak viruses (SARS-CoV-1, MERS-CoV, and SARS-CoV-2) and various chimeric coronaviruses as positive controls. We identify genomic regions evolving under distinct selective pressures. We also trace possible signs of human-made manipulations using metrics such as synonymous and non-synonymous mutation rates, codon usage and insertion patterns. Our pipeline appears to efficiently detect synthetic sequence optimization or genome re-encoding, but does not identify chimeras of natural viruses. Unlike in positive controls, with our method no signal of human-made manipulation is detected in SARS-CoV-1, MERS-CoV, nor SARS-CoV-2.