ESMRank reveals a transferable axis of protein mutational constraint from overlapping variant effect assays
ESMRank reveals a transferable axis of protein mutational constraint from overlapping variant effect assays
Arnese, R.; Gambardella, G.
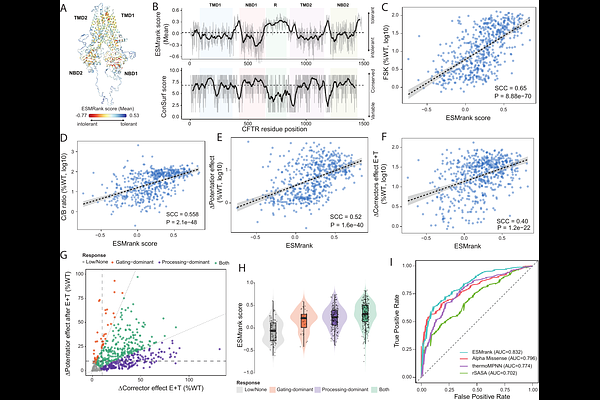
AbstractProteome-wide interpretation of missense variation is constrained not only by predictive model performance but also by the absence of principled methods to reconcile heterogeneous multiplexed assays of variant effect (MAVEs) into a unified representation of mutational constraint. We show that redundancy among partially overlapping deep mutational scanning experiments encodes a reproducible ordinal signal that can be recovered despite differences in assay scale and readout. We introduce variant soundness, an overlap-aware framework that aligns within-assay rankings and aggregates them across experiments to derive an assay-agnostic, within-protein measure of mutational tolerance. Applying this approach to about 1,100 MAVEdb score sets spanning >2M variants reveals a coherent constraint landscape enriched for structural stability determinants, including residue burial, packing perturbation magnitude, and domain architecture. By aligning learning objectives with this intrinsic ordering, we develop ESMRank, a sequence-based learning-to-rank predictor integrating protein language model representations with physicochemical descriptors. Under strict protein-level partitioning, ESMRank outperforms widely used stability and fitness predictors across the Human Domainome, ProteinGym stability assays, and VariBench folding kinetics. Without clinical supervision, the reconstructed constraint axis is enriched for ClinVar pathogenic variants and stratifies genes by mechanistic disease classes. In CFTR, predicted constraint tracks folding efficiency, channel activity, and pharmacological rescue. These findings establish experimental overlap as a scalable resource for extracting transferable mutational ordering and for building mechanistically interpretable, proteome-wide variant effect predictors.