Genotype imputation in F2 crosses of inbred lines
Genotype imputation in F2 crosses of inbred lines
Pierotti, S.; Welz, B.; Fitzgerald, T.; Wittbrodt, J.; Birney, E.
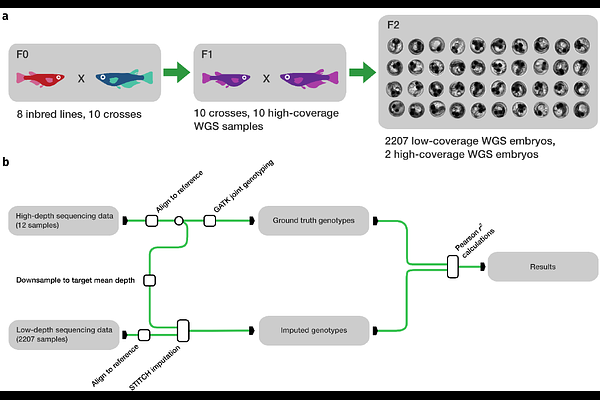
AbstractMotivation: Crosses among inbred lines are a fundamental tool for the discovery of genetic loci associated with phenotypes of interest. In organisms for which large reference panels or SNP chips are not available, imputation from low-pass whole-genome sequencing is an effective method for obtaining genotype data from a large number of individuals. To date, a structured analysis of the conditions required for optimal genotype imputation has not been performed. Results: We report a systematic exploration of the effect of several design variables on imputation performance in F2 crosses of inbred medaka lines. We determined that, depending on the number of samples, imputation performance reaches a plateau when increasing the per-sample sequencing coverage. We also systematically explored the tradeoffs between cost, imputation accuracy, and sample numbers. We developed a computational pipeline to streamline the process, enabling other researchers to perform a similar cost-benefit analysis on their population of interest.