Metagenome profiling and containment estimation through abundance-corrected k-mer sketching with sylph
Metagenome profiling and containment estimation through abundance-corrected k-mer sketching with sylph
Shaw, J.; Yu, Y. W.
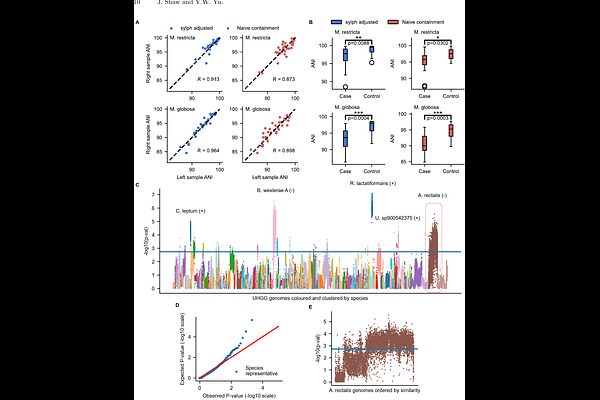
AbstractProfiling metagenomes against databases allows for the detection and quantification of microbes, even at low abundances where assembly is not possible. We introduce sylph, a metagenome profiler that estimates metagenome-genome average nucleotide identity (ANI) through zero-inflated Poisson k-mer statistics, enabling ANI-based taxa detection. Sylph is the most accurate method on the CAMI2 marine dataset, and compared to Kraken2 for multi-sample profiling, sylph takes 10x less CPU time and uses 30X less memory. Sylph\'s ANI estimates provide an orthogonal signal to abundance, enabling an ANI-based metagenome-wide association study for Parkinson\'s disease against 289,323 genomes, confirming known butyrate-PD associations at the strain level. Sylph takes < 1 minute and 16 GB of RAM to profile against 85,205 prokaryotic and 2,917,521 viral genomes, detecting 30X more viral sequences in the human gut compared to RefSeq. Sylph offers precise, efficient profiling with accurate ANI estimation for even low-coverage genomes. Code for sylph: https://github.com/bluenote-1577/sylph