Next-generation all-in-one CRISPR/Cas9 multiply-edited CD30CAR-T cells:Potency despite risk of translocations
Next-generation all-in-one CRISPR/Cas9 multiply-edited CD30CAR-T cells:Potency despite risk of translocations
Kleid, J.-M.; Damrat, M.; Darguzyte, M.; Rhiel, M.; Stumpf, N. E.; Kleitke, T.; Ammann, S.; Cornu, T. I.; Khan, F.; Wollmann, T.; Borchmann, S.; Scheid, C.; Moraes, C.; Riet, T.; Awerkiew, S.; Ullrich, L.; Gathof, B.; Klawonn, F.; Eiz-Vesper, B.; Wagner, D. L.; Huebel, K.; Ullrich, R.; Bornhaeuser, M.; Cathomen, T.; Stripecke, R.
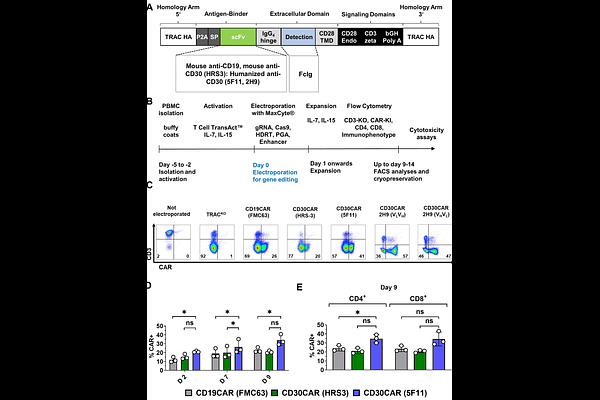
AbstractBackground: Chimeric antigen receptor (CAR)-T cells are therapeutic breakthroughs against advanced non-Hodgkin lymphomas and myelomas. On the other hand, no CAR-T cell product has been so far clinically approved for therapy of Hodgkin Lymphoma (HL), T cell lymphoma (TCL), or Epstein-Barr-Virus (EBV)-associated lymphoproliferative diseases (EBV-LPDs). CD30 (TNFRSF8) is commonly expressed on HL and on subsets of TCL and EBV-LPDs. CD30CAR-T cells generated via transduction with viral vectors have been tested in clinical trials, showing overall good responses against HL. CAR-T cells produced entirely with locus-specific gene editing methods are emerging as attractive next-generation engineered cell products for ease of multiple seamless cell modifications. Methods: Using CRISPR/Cas9-mediated techniques, we optimized homology-directed repair templates (HDRTs) and performed all-in-one multiplex editing to knock-in (KI) CD30CAR within the TCR constant (TRAC) locus and to simultaneously knock-out (KO) PD-1 or/and {beta}2M. CD30CAR-T cells were tested in CD30+ cell models of HL, TCL, and EBV-LPDs. Results: We compared mouse versus human anti-CD30 scFv designs in HDRTs incorporating TRAC homology arms, FcIg spacer/detection domain, and CD28 / CD3{zeta} signaling domains. We obtained an average of 30% TRACKICD30CAR-T cells and efficient in vitro cytotoxicity with CD30+ cell targets. CARs incorporating the high-affinity humanized 5F11 scFv showed the highest CAR expression, and the editing templates were further modified to incorporate a truncated CD34 (tCD34) spacer/detection domain. 5F11-CD30CAR-tCD34-T cells showed high CAR-KI rates (approx. 50-80% 12-14 days after editing) and potency in vitro and in vivo. Subsequently, we tested all-in-one CAR KI with additional KOs by co-electroporation of guide RNAs (gRNAs) targeting the genes encoding PD-1 or /and {beta}2M to improve function and allow for improved cell persistence in allogeneic recipients, respectively. Compared with CD30CAR-T cells, CD30CAR-{beta}2MKO-T cells were similarly viable and functional and showed low risk of translocations. PD1KO enabled CD30CAR-T cells to produce higher levels of cytotoxic features upon exposure to targets. However, simultaneous {beta}2MKO and PD-1KO compromised the expansion capacity of CD30CAR-T cells and resulted in detectable translocations. Conclusions: Non-virally engineered 5F11-CD30CAR-T cells represent a novel cell therapy modality against CD30+ lymphomas. Multiplex editing remains to be optimized to avoid unwanted genomic alterations and chromosomal translocations.