BinDash 2.0: New MinHash Scheme Allows Ultra-fast and Accurate Genome Search and Comparisons
BinDash 2.0: New MinHash Scheme Allows Ultra-fast and Accurate Genome Search and Comparisons
Zhao, J.; Zhao, X.; Pierre-Both, J.; Konstantinidis, K. T.
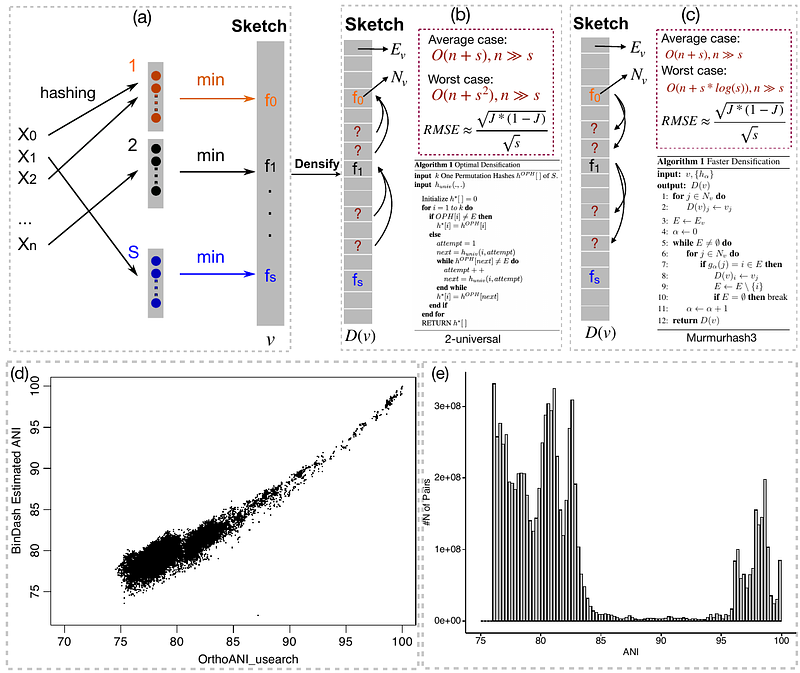
AbstractMotivation: Comparing large number of genomes in term of their genomic distance is becoming more and more challenging because there is an increasing number of microbial genomes deposited in public databases. Nowadays, we may need to estimate pairwise distances between millions or even billions of genomes. Few softwares can perform such comparisons efficiently. Results: Here we update the multi-threaded software BinDash by implementing several new MinHash algorithms and computational optimization (e.g. Simple Instruction Multiple Data, SIMD) for ultra-fast and accurate genome search and comparisons at trillion scale. That is, we implemented b-bit one-permutation rolling MinHash with optimal/faster densification with SIMD. Now with BinDash 2, we can perform 0.1 trillion (or ~10^11) pairs of genome comparisons in about 1.8 hours on a descent computer cluster or several hours on personal laptops, a ~50% or more improvement over original version. The ANI (average nucleotide identity) estimated by BinDash is well correlated with other accurate but much slower ANI estimators such as FastANI or alignment-based ANI. In line with the findings from comparing 90K genomes (~10^9 comparisons) via FastANI, the 85% ~ 95% ANI gap is consistent in our study of ~10^11 prokaryotic genome comparisons via BinDash2, which indicates fundamental ecological and evolutionary forces keeping species-like unit (e.g., > 95% ANI) together.