Characterization of a novel mouse model of Dopamine Transporter Deficiency Syndrome and pharmacological therapeutic strategies
Characterization of a novel mouse model of Dopamine Transporter Deficiency Syndrome and pharmacological therapeutic strategies
Russo, E.; Rezayof, A.; Wallace, C. W.; Williams, E. Q.; Beerepoot, P.; Milenkovic, M.; Novalen, M.; Blundell, A.; Lipina, T. V.; Locke, J.; Christian, R.; Finnie, P. S. B.; Edgar, L. J.; Tyndale, R.; Watkins-Chow, D. E.; Ramsey, A. J.; Jones, S. R.; Salahpour, A.
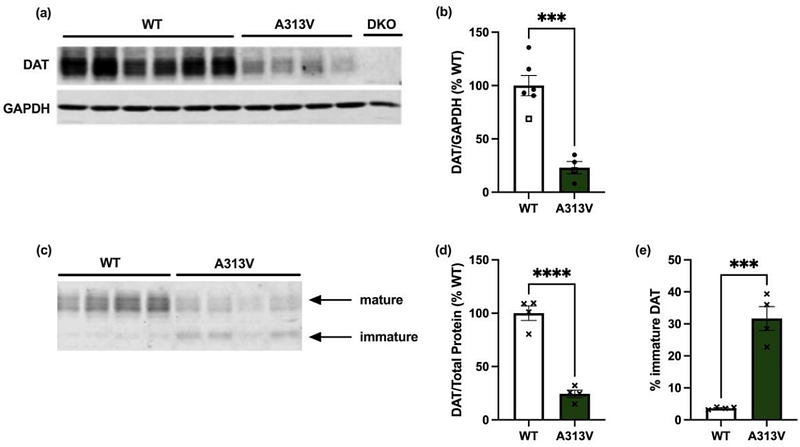
AbstractThe dopamine transporter (DAT) is an essential protein in the maintenance of dopamine homeostasis. Single amino acid changes in the DAT gene SLC6A3 can be sufficient to induce disease, such as Dopamine Transporter Deficiency Syndrome (DTDS). DTDS-associated variants are posited to cause DAT protein misfolding, retention in the endoplasmic reticulum, and a reduction in DAT at the cell surface. In turn, proper dopaminergic regulation is lost. Current treatments for DTDS are largely ineffective and improved therapeutic options are greatly needed. To this end, we have created a novel mouse model of DTDS harboring the A313V knock-in DAT variant, a proxy for the DTDS-causing A314V human variant. A313V knock-in DAT mice are hyperactive, have increased striatal tissue content of dopamine and its metabolites, and impaired dopamine uptake. FDA approved compounds alpha-methyl-para-tyrosine ([a]MPT) and amphetamine (AMPH) ameliorate hyperactivity in this mouse model. Moreover, [a]MPT may be a disease-modifying treatment by addressing the hyperdopaminergic tone underlying this hyperactivity. In contrast, noribogaine, a pharmacological chaperone for DAT in cells, is unable to rescue DAT expression. These findings show that the A313V knock-in DAT variant mice recapitulate several defining phenotypes seen in patients with DTDS, and provide evidence for two novel treatments for the disease.