Single cell Correlation Analysis (SCA): Identifying self-renewing subpopulation of human acute myeloid leukemia stem cells using single cell RNA sequencing analysis
Single cell Correlation Analysis (SCA): Identifying self-renewing subpopulation of human acute myeloid leukemia stem cells using single cell RNA sequencing analysis
Lee, Y.; Wang, W.; Starr, T. K.; Noble-Orcutt, K. E.; Myers, C. L.; Sachs, Z.
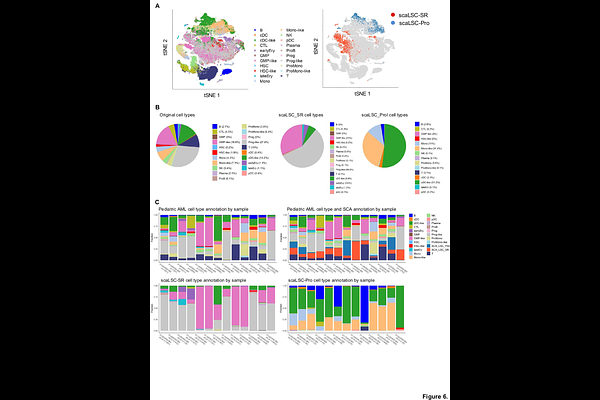
AbstractLeukemia stem cells (LSCs), a rare and self-renewing subpopulation, drive Acute myeloid leukemia (AML) relapse and therapy resistance. While single-cell gene expression profiling (scGEP) offers high-resolution insights into LSC biology, existing computational tools rely on relative and arbitrary similarity metrics. This limits their ability to definitively identify true biological cell identities or compare rare cell populations across independent datasets. To overcome these limitations, we developed Single cell Correlation Analysis (SCA), a novel computational method that utilizes a permutation-based false discovery rate (FDR) framework and common background datasets to establish standardized, statistically rigorous thresholds of similarity. We applied SCA to query human AML scRNA-seq datasets using an experimentally validated murine scGEP of LSC self-renewal as our reference. SCA demonstrated superior specificity and precision, maintaining low false positive rates compared to existing reference-based annotation tools. Using SCA, we successfully identified cells expressing a conserved self-renewal program (scaLSC-SR) in both adult and pediatric human AML samples. These scaLSC-SR cells share classical LSC immunophenotypic markers (e.g., CD34, CD96, CD200) and exhibit transcriptomic hallmarks of LSC biology, including evasion of apoptosis and immune responses. Furthermore, this self-renewal program is significantly enriched in poor-risk genetic subtypes, specifically TP53 and NRAS mutated AML. Finally, we derived a 28-gene signature (LSC-SR28) from human AML patients that accurately captures LSC stemness and is highly prognostic across multiple independent clinical cohorts. SCA provides a robust, statistically principled framework for identifying rare and biologically meaningful cell populations across heterogeneous single-cell datasets. By successfully identifying a functional self-renewal profile to human AML, SCA reveals conserved, clinically relevant LSC populations that drive relapse and therapy resistance across different age groups.