Adaptive Integration of Heterogeneous Foundation Models to Find Histologically Predictable Genes in Breast Cancer
Adaptive Integration of Heterogeneous Foundation Models to Find Histologically Predictable Genes in Breast Cancer
Nguyen, H.; Li, C.; Peng, C.; Simpson, P.; Ye, N.; Nguyen, Q.
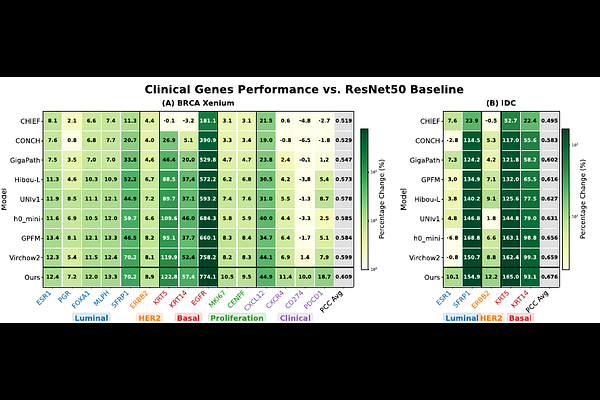
AbstractFoundation models for computational pathology have rapidly emerged as powerful tools for extracting rich biological and morphological representations from histopathology images. However, variations in model architecture, pre-training data, and optimization objectives often lead to task-dependent performance, rather than universal generalization. As a result, effective strategies for integrating their complementary strengths are essential to fully realize the potential of foundation models for robust histopathology analysis. Meanwhile, recent breakthroughs such as spatial transcriptomics provide an unprecedented opportunity to integrate genetic and histopathology information from the same patient sample, thereby maximizing both molecular and anatomical pathology insights. Specifically, each model's embedding is first mapped to gene-level predictions via a dedicated prediction head, enabling model-specific feature utilization. A lightweight weighting network then adaptively aggregates these predictions to produce a unified and robust output at gene and spatial location levels. Across multiple spatial transcriptomics datasets, our approach consistently outperforms both individual foundation models and classical ensembling methods. Focusing on breast cancer, we observe substantial gains in prediction accuracy for clinically relevant PAM50 subtype markers and drug-target genes. Moreover, the proposed framework improves interpretability by revealing model-specific contributions and specialization at the gene level. Overall, our work presents an effective solution to integrating multiple foundation models for enhancing the genetic analyses of histopathology images.