Identifying non-coding variant effects at scale via machine learning models of cis-regulatory reporter assays
Identifying non-coding variant effects at scale via machine learning models of cis-regulatory reporter assays
Butts, J. C.; Rong, S.; Gosai, S. J.; Castro, R. I.; Noon, M.; Adeniran, K.; Ghosh, R.; Sabeti, P. C.; Tewhey, R.; Reilly, S. K.
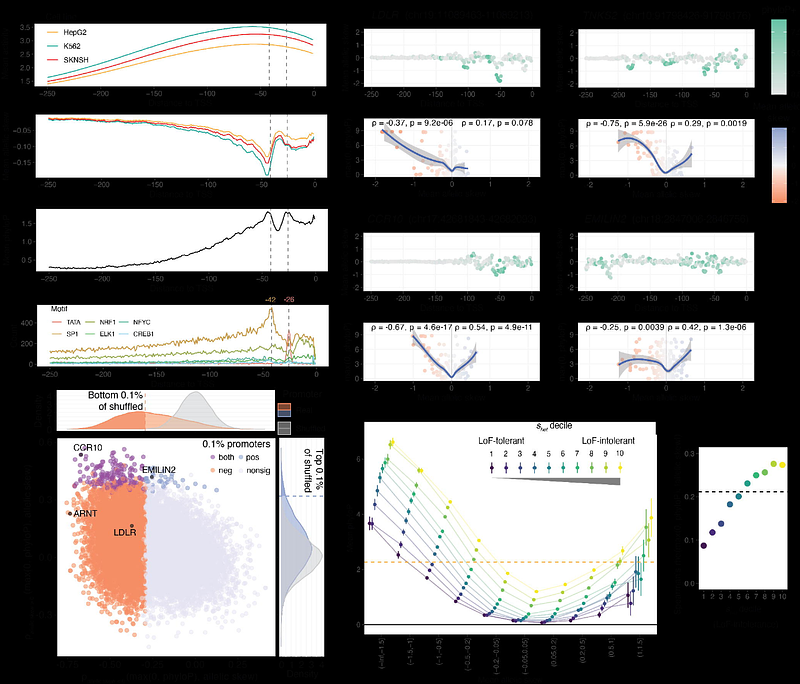
AbstractThe inability to interpret the functional impact of non-coding variants has been a major impediment in the promise of precision medicine. While high-throughput experimental approaches such as Massively Parallel Reporter Assays (MPRAs) have made major progress in identifying causal variants and their underlying molecular mechanisms, these tools cannot exhaustively measure variant effects genome-wide. Here we present MPAC, an ensemble of machine-learning models trained on MPRA data that provides accurate and scalable prediction of the cis-regulatory impact of non-coding variants. Using MPAC we predict allelic effects for 575M single nucleotide variants (SNVs) across diverse applications, including complex trait genetics, clinical and tumor sequencing, evolutionary analyses, and saturation mutagenesis. We find MPAC predictions match the performance of empirical MPRAs in identifying causal complex trait-associated alleles. We demonstrate the utility of MPAC by applying it to ClinVar, identifying non-coding pathogenic variation with higher accuracy than other sequence-to-function models. We also nominate 1,892 candidate non-coding cancer drivers by predicting the functional effects of somatic SNVs in the COSMIC database. Next, we evaluate population-level genetic variation by predicting effects for all 514M non-coding SNVs in gnomAD, quantifying the relationship between regulatory function and evolutionary constraint. Finally, we generate prospective functional maps using in-silico saturation mutagenesis across 18,658 human promoters, observing widespread selection against variants predicted to disrupt promoter activity. Collectively, this study establishes the value of non-coding functional predictions and provides a comprehensive, publicly available resource for variant interpretation.