Do AI Models for Protein Structure Prediction Get Electrostatics Right?
Do AI Models for Protein Structure Prediction Get Electrostatics Right?
Makhatadze, G. I.
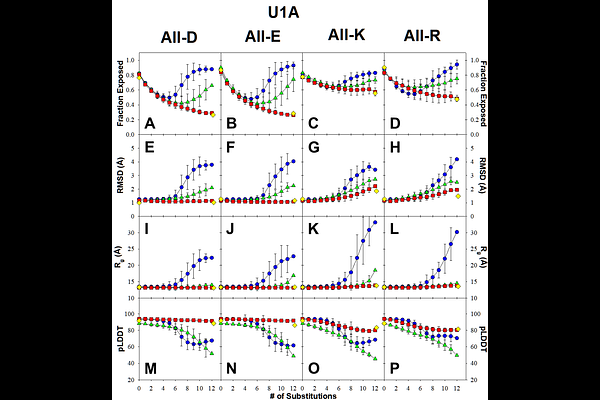
AbstractA variant of the U1A protein containing four substitutions to ionizable residues was generated serendipitously due to a miscommunication. Biophysical measurements show that this variant has at least twice as much helical structure as the wild-type U1A and is trimeric in solution, in contrast to the monomeric wild type. In sharp contrast, structures predicted by deep-learning AI tools (AlphaFold2 and RoseTTAFold2) and transformer-based tools (OmegaFold and ESMFold) are all highly similar to the wild-type U1A (backbone RMSD < 1 [A]). Even more surprising, two of the substituted ionizable residues are predicted to be fully buried in the non-polar core of the protein, an outcome that contradicts well-established physico-chemical principles, as ionizable residues are normally located on the protein surface. To explore this effect further, we generated sequences containing up to all twelve residues that make up the non-polar core of U1A. Across thousands of sequences, and depending on the AI model used, the majority of predicted structures contained fully buried ionizable residues while still maintaining the overall U1A fold. We then examined two additional proteins of comparable size, acylphosphatase and the de novo designed TOP7 fold, and observed the same phenomenon: AI models frequently predicted structures with buried ionizable residues that nevertheless retained the parent fold. When these AI-predicted structures were subjected to short (50 ns) molecular dynamics simulations using physics-based force fields such as CHARMM or AMBER, the structures rapidly relaxed into ensembles that exposed ionizable residues. We conclude that while AI-based structure prediction tools perform extremely well on naturally occurring sequences, they do not reliably encode the physico-chemical principles governing the placement of ionizable residues. A straightforward remedy is to include a brief molecular dynamics simulation as a final validation step for AI-generated structures.