The role of liprin-α1 phosphorylation in its liquid-liquid phase separation: regulation by PPP2R5D/PP2A holoenzyme
The role of liprin-α1 phosphorylation in its liquid-liquid phase separation: regulation by PPP2R5D/PP2A holoenzyme
Mayer, A.; Derua, R.; Spahn, E.; Verbinnen, I.; Zhang, Y.; Wadzinski, B.; Swingle, M.; Honkanen, R.; Janssens, V.; Xia, H.
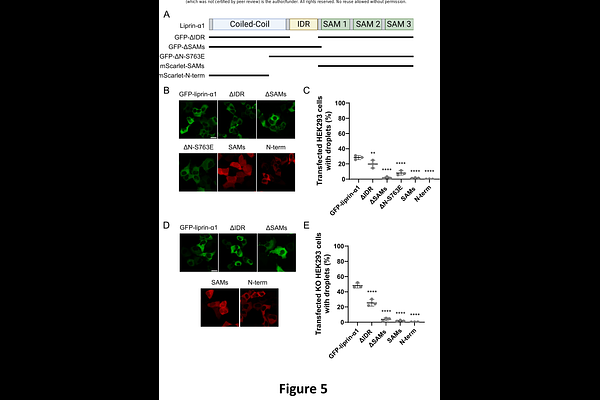
AbstractLiprin-alpha1 is a widely expressed scaffolding protein responsible for regulating cellular processes such as focal adhesion, cell motility, and synaptic transmission. Liprin-alpha1 interacts with many proteins including ELKS, GIT1, liprin-beta, and LAR-family receptor tyrosine protein phosphatase. Through these protein-protein interactions, liprin-alpha1 assemble large higher-order molecular complexes; however, the regulation of this complex assembly/disassembly is unknown. Liquid-liquid phase separation (LLPS) is a process that concentrates proteins within cellular nano-domains to facilitate efficient spatiotemporal signaling in response to signaling cascades. While there is no report that liprin-alpha1 spontaneously undergoes LLPS, we found that GFP-liprin-alpha1 expressed in HEK293 cells occasionally forms droplet-like condensates. MS-based interactomics identified Protein Phosphatase 2A (PP2A)/B56delta (PPP2R5D) trimers as specific interaction partners of liprin-alpha1 through a canonical Short Linear Interaction Motif (SLiM) in its N-terminal dimerization domain. Mutations of this SLiM nearly abolished PP2A interaction and resulted in significantly increased LLPS. GFP-liprin-alpha1 showed significantly increased droplet formation in HEK293 cells devoid of B56delta (PPP2R5D knockout), suggesting that PPP2R5D/PP2A holoenzyme inhibits liprin-alpha1 LLPS. Guided by reported liprin-alpha1 Ser/Thr phosphorylation sites, we found liprin-alpha1 phospho-mimetic mutant at serine 763 (S763E) is sufficient to drive its LLPS. Domain mapping studies of liprin-alpha1 indicated that the intrinsically disordered region, the N-terminal dimerization domain, and the SAM domains are all necessary for liprin-alpha1 LLPS. Finally, expression of p.E420K, a human PPP2R5D variant causing Houge-Janssens Syndrome type 1(also known as Jordan Syndrome), significantly compromised suppression of liprin-alpha1 LLPS. Our work identified B56delta-PP2A holoenzyme as an inhibitor of liprin-alpha LLPS via regulation at multiple phosphorylation sites.