Optimizing Cost-Effective Gene Expression Phenotyping Approaches in Cattle Using 3' mRNA Sequencing


Optimizing Cost-Effective Gene Expression Phenotyping Approaches in Cattle Using 3' mRNA Sequencing
Mohamed, R. I.; Ault-Seay, T. B.; Moisa, S.; Beever, J. E.; Rius, A. G.; Rowan, T. N.
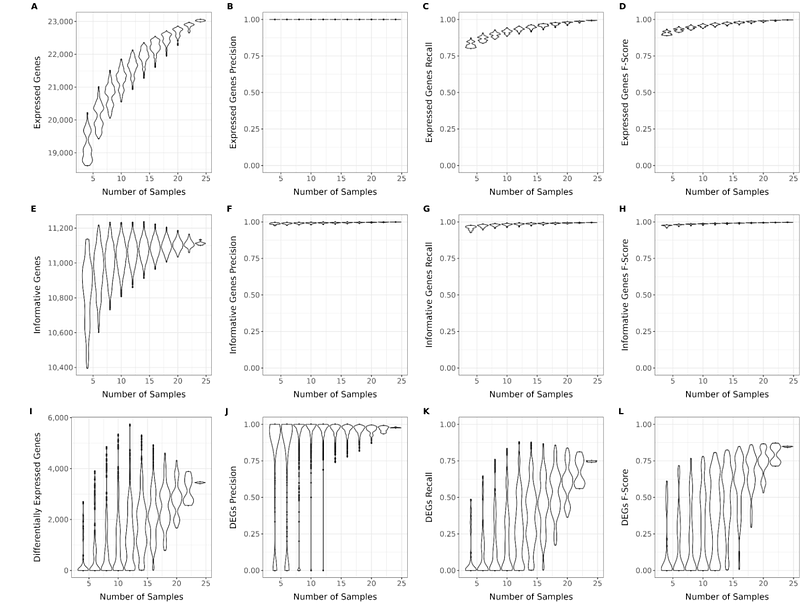
AbstractGenetic and genomic selection programs require large numbers of phenotypes observed for animals in shared environments. Direct measurements of phenotypes like meat quality, methane emission, and disease susceptibility are difficult and expensive to measure at scale but are critically important to livestock production. Our work leans on our understanding of the Central Dogma of molecular genetics to leverage molecular intermediates as cheaply-measured proxies of organism-level phenotypes. The rapidly declining cost of next-generation sequencing presents opportunities for population-level molecular phenotyping. While the cost of whole transcriptome sequencing has declined recently, its required sequencing depth still makes it an expensive choice for wide-scale molecular phenotyping. We aim to optimize 3\' mRNA sequencing (3\' mRNA-Seq) approaches for collecting cost-effective proxy molecular phenotypes for cattle from easy-to-collect tissue samples (i.e., whole blood). We used matched 3\' mRNA-Seq samples for 15 Holstein male calves in a heat stress trail to identify the 1) best library preparation kit (Takara SMART-Seq v4 3\' DE and Lexogen QuantSeq) and 2) optimal sequencing depth (0.5 to 20 million reads/sample) to capture gene expression phenotypes most cost-effectively. Takara SMART-Seq v4 3\' DE outperformed Lexogen QuantSeq libraries across all metrics: number of quality reads, expressed genes, informative genes, differentially expressed genes, and 3\' biased intragenic variants. Serial downsampling analyses identified that as few as 8.0 million reads per sample could effectively capture most of the between-sample variation in gene expression. However, progressively more reads did provide marginal increases in recall across metrics. These 3\' mRNA-Seq reads can also capture animal genotypes that could be used as the basis for downstream imputation. The 10 million read downsampled groups called an average of 104,386 SNPs and 20,131 INDELs, many of which segregate at moderate minor allele frequencies in the population. This work demonstrates that 3\' mRNA-Seq with Takara SMART-Seq v4 3\' DE can provide an incredibly cost-effective (<$25/sample) approach to quantifying molecular phenotypes (gene expression) while discovering sufficient variation for use in genotype imputation. Ongoing work is evaluating the accuracy of imputation and the ability of much larger datasets to predict individual animal phenotypes.