Resolving the full set of human polymorphic inversions and other complex variants from ultra-long read data
Resolving the full set of human polymorphic inversions and other complex variants from ultra-long read data
Moreira-Pinhal, R.; Karakostis, K.; Yakymenko, I.; Conchillo, O.; Diaz-Ros, M.; Santos, A.; Senar, M. A.; Martinez-Urtaza, J.; Puig, M.; Caceres, M.
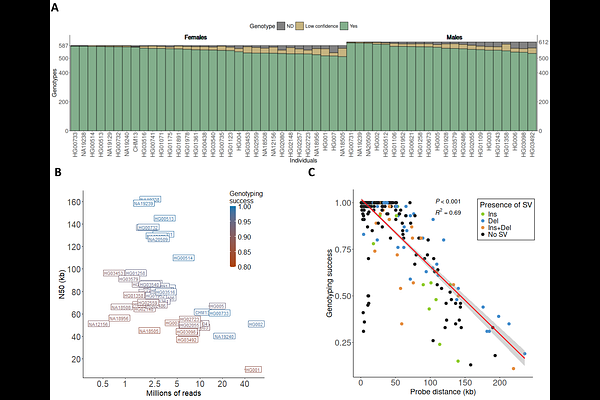
AbstractInversions are a unique type of balanced structural variants (SVs) with important consequences in multiple organisms. However, despite considerable effort, this and other complex SVs remain poorly characterized due to the presence of large repeats. New techniques are finally allowing us to identify the full spectrum of human inversions, but the number of individuals analyzed is still quite limited. Here, we take advantage of Oxford Nanopore Technologies (ONT) long reads to characterize an exhaustive catalogue of 612 candidate inversions between 197 bp and 4.4 Mb of length and flanked by <190-kb long inverted repeats (IRs). For that, we developed a bioinformatic package to identify inversion alleles reliably from long read data. Next, using a combination of different DNA extraction, library preparation, and ONT sequencing protocols, we showed that ultra-long reads (50-100 kb) and adaptive sampling are an efficient method to detect most human inversions. Lastly, by analyzing ONT data from 54 diverse individuals, 87-99% of the inversions could be genotyped in each sample, depending mainly on read and IR length and genome coverage. Both orientations were observed for 155 of the analyzed regions (frequency 0.01-0.49), which multiplies by three the polymorphic IR-mediated inversions studied in detail so far. Moreover, we found more than 300 additional independent SVs in the studied regions and resolved several complex rearrangements. Our work therefore provides an accurate benchmark of those inversions that typically escape most analyses, improving existing resources, such as the Pangenome. In addition, it demonstrates the potential of nanopore sequencing to determine the functional impact of missing human genomic variation.