A Tunable, Ultrasensitive Threshold in Enzymatic Activity Governs the DNA Methylation Landscape


A Tunable, Ultrasensitive Threshold in Enzymatic Activity Governs the DNA Methylation Landscape
Bonsu, K. A.; Trinh, A.; Downing, T. L.; Read, E. L.
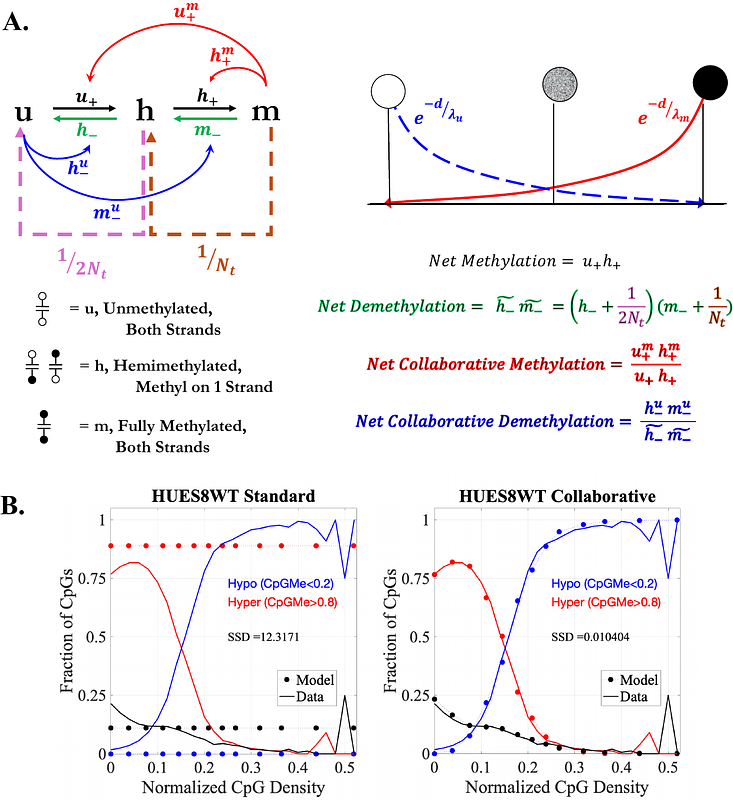
AbstractDNA methylation is a widely studied epigenetic mark, affecting gene expression and cellular function at multiple levels. DNA methylation in the mammalian genome occurs primarily at cytosine-phosphate-guanine (CpG) dinucleotides, and patterning of the methylation landscape (i.e., the presence or absence of CpG methylation at a given genomic location) exhibits a generally bimodal distribution. Although much is known about the enzymatic writers and erasers of CpG methylation, it is not fully understood how these enzymes, along with genetic, chromatin, and regulatory factors, control the genome wide methylation landscape. In this study, methylation is analyzed at annotated CpG Islands (CGIs) and independent CpGs as a function of their proximity to other CpG substrates. Analysis is aided by a computationally efficient stochastic mathematical model of methylation dynamics, enabling parameterization from data. We find that methylation exhibits a switch-like dependence on local CpG density. The threshold and steepness of the switch is modified in cell lines in which key enzymes are knocked out. Modeling further elucidates how enzymatic parameters, including catalytic rates and lengthscales of inter-CpG interaction, tune the properties of the switch. Together, the results support a model in which competition between opposing TET1-3 demethylating enzymes and DNA methyltransferases (DNMT3A/B) results in an ultrasensitive switch, analogous to the protein phosphorylation switch (termed \"zero-order ultrasensitivity\") proposed by Goldbeter and Koshland. Our study provides insight to the mechanisms underlying establishment and maintenance of bimodal DNA methylation landscapes, and further provides a flexible pipeline for gleaning molecular insights to the cellular methylation machinery across cell-specific, epigenomic datasets.