Longitudinal study of the udder microbiome of Norwegian red dairy cows using metataxonomic and shotgun metagenomic approaches: Insights into pathogen-driven microbial adaptation and succession
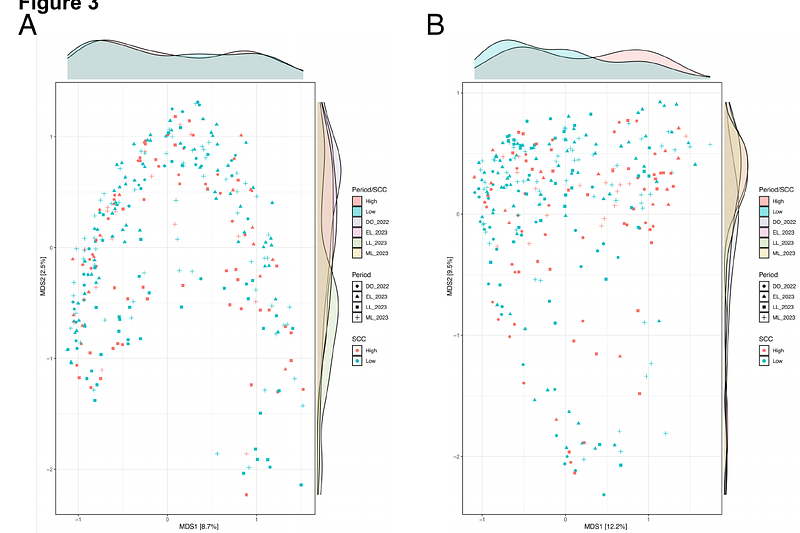
Longitudinal study of the udder microbiome of Norwegian red dairy cows using metataxonomic and shotgun metagenomic approaches: Insights into pathogen-driven microbial adaptation and succession
Da Silva Duarte, V.; Franklin, F.; Krysmann, A. M.; Porcellato, D.
AbstractBovine mastitis remains the most significant disease affecting dairy herds globally, driven by its multi etiological nature and the complex dynamics of udder immunity and infection. While research addressing the microbial and immunological aspects of the bovine udder is limited, optimizing the udder microbiome has emerged as a promising strategy for preventing mastitis. This longitudinal study aimed to investigate the udder microbiome throughout lactation using both metataxonomic and shotgun metagenomic approaches, including analysis at the metagenome assembled genome (MAG) level. The use of such methodologies provide a deeper understanding of the microbial composition and dynamics within the udder, bridging critical gaps in knowledge and revealing potential interactions between the microbiota and host. Milk samples were collected from 342 individual quarters of 24 Norwegian Red dairy cows. Significant variations in somatic cell count and microbiota composition were observed across lactation stages. Quarters with low somatic cell count (< 100,000 cells/mL) were notably higher during early lactation (80%) and mid-lactation (78.9%) compared to dry-off (53.1%) and late lactation (53%), with high somatic cell count (> 100,000 cells/mL) observed in 20 and 47% of samples. Diversity indices based on Shannon and Simpson metrics indicated significant effects of lactation stage, somatic cell count, and individual animal variability on microbial diversity. PERMANOVA analyses confirmed that individual animal variability (15.73%) and lactation period (5.52%) were the strongest factors influencing microbiota structure, with dysbiosis linked to mastitis-causing pathogens contributing 7.17% of the variance. Key pathogens identified included E. faecalis, S. aureus, S. uberis, and S. chromogenes, with persistent infections observed for S. aureus and S. chromogenes, but not S. uberis. Samples with low somatic cell count were enriched in beneficial genera such as Corynebacterium, Bradyrhizobium, and Lactococcus, while Staphylococcus predominated in milk samples with high somatic cell count. Dimensionality reduction with tSNE and integration with culturomics enhanced milk microbiota classification, providing novel insights into udder microbiota dynamics and their role in bovine mastitis. Metagenomic shotgun sequencing revealed pathogen-specific metabolic signatures in the bovine mammary gland, identifying 289 MetaCyc pathways. Genome-centric analysis reconstructed 142 metagenome-assembled genomes, including 26 from co-assembly and 116 from individual assembly. Multi-locus sequence typing, virulence factors, and antimicrobial resistance gene profiling provided insights into pathogen adaptation and persistence in the bovine mammary gland. Notably, 27 bacteriocin gene clusters and 322 biosynthetic gene clusters were predicted using genome mining tools. Our findings contribute to the establishment of pathogen based therapies and enhance our understanding of the milk microbiome, which remains far from fully characterized. Such insights are vital for developing effective strategies to combat mastitis and improve dairy cattle health.