KinConfBench: A Curated Benchmark for Cofolding Models on Kinase Conformational States
KinConfBench: A Curated Benchmark for Cofolding Models on Kinase Conformational States
Sun, K.; Head-Gordon, T.
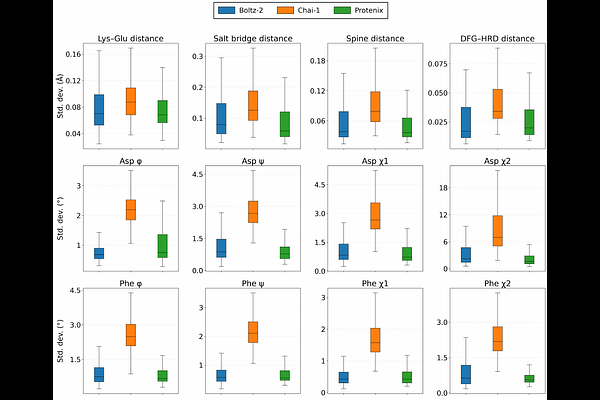
AbstractProtein kinases are critical drug targets, requiring therapeutics that can modulate their active and inactive conformational states. While cofolding models can generate global folds directly from kinase sequences and ligand SMILES strings, these models have not yet been tested on their ability to recover ligand induced-fit conformational states of the kinase proteins. Here, we introduce KinConfBench, a curated benchmark of 2,225 high-quality human kinase chains to evaluate the ability of three state-of-the-art cofolding models---Boltz-2, Chai-1, and Protenix---to recover both canonical and rare conformational states. We show that geometric success metrics of a ligand pose in the active site does not correlate strongly with the correct kinase conformational state, motivating a new set of dynamical benchmarks for assessing cofolding models. While all three cofolding models achieve ~65-75% prediction accuracy for kinase conformational classification, they exhibit severe mode collapse when performing multiple inferences, show negligible structural diversity in sampling induced-fit motions, and display a prevalent ``apo-drift'' in which all three cofolding models predominately predict the kinase to be in its ligand-free state. Our results highlight that capturing ligand-induced protein conformational diversity, not just geometric fit, is critical for next-generation structure-based drug discovery.