Cell modeling and rescue of a novel non-coding genetic cause of Glycogen Storage Disease IX
Cell modeling and rescue of a novel non-coding genetic cause of Glycogen Storage Disease IX
Iyengar, A. K.; Zou, X.; Dai, J.; Francis, R. A.; Safi, A.; Patterson, K.; Koch, R. L.; Clarke, S.; Beaman, M. M.; Chong, J. X.; Bamshad, M. J.; Majoros, W. H.; Rehder, R. C.; Bali, D. S.; Allen, A. S.; Crawford, G. E.; Kishnani, P. S.; Reddy, T. E.
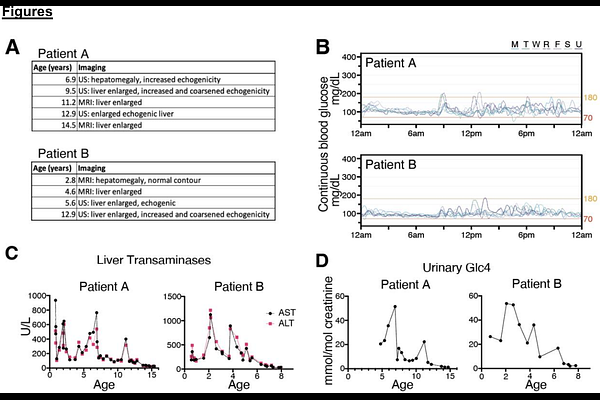
AbstractDelayed diagnosis of Mendelian disease substantially prevents early therapeutic intervention that could improve symptoms and prognosis. One major contributing challenge is the functional interpretation of non-coding variants that cause disease by altering splicing and/or gene expression. We identified two siblings with glycogen storage disease (GSD) type IX {gamma}2, both of whom had a classic clinical presentation, enzyme deficiency, and a known pathogenic splice acceptor variant on one allele of PHKG2. Despite the autosomal recessive nature of the disease, no variant on the second allele was identified by gene panel sequencing. To identify a potential missing second pathogenic variant, we completed whole genome sequencing (WGS) and detected putative deep intronic splicing variant in PHKG2 in both siblings. We confirmed the functional splicing effects of this variant using short-read and long-read RNA-seq on patient blood and a HEK293T cell model in which we installed the variant using CRISPR editing. Using the cell model, we demonstrated multiple biochemical and cellular impacts that are consistent with GSD IX {gamma}2, and a reversal of aberrant splicing using antisense splice-switching oligonucleotides. In doing so, we demonstrate a novel and robust pathway for detecting, validating, and reversing the impacts of novel non-coding causes of rare disease.