Optimizing mouse metatranscriptome profiling by selective removal of redundant nucleic acid sequences
Optimizing mouse metatranscriptome profiling by selective removal of redundant nucleic acid sequences
Kuersten, S.; Roos, M.; Tan, A.; Bunga, S.; Skola, D.; Conrad, R.; Maissy, E. S.; Richter, A.; Desplats, P.; Whittaker, D. S.; Zarrinpar, A.
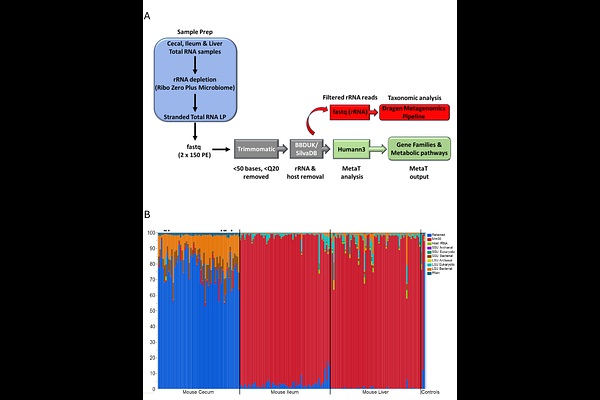
AbstractMetatranscriptome (MetaT) sequencing is a critical tool for profiling the dynamic metabolic functions of microbiomes. In addition to taxonomic information, MetaT also provides real-time gene expression data of both host and microbial populations, thus permitting authentic quantification of the functional (enzymatic) output of the microbiome and its host. The main challenge to effective and accurate MetaT analysis is the removal of highly abundant rRNA transcripts from these complex mixtures of microbes, which can number in the thousands of individual species. Regardless of methodology for rRNA depletion, the design of rRNA removal probes based solely upon taxonomic content of the microbiome typically requires very large numbers of individual probes, making this approach complex to commercially manufacture, costly, and frequently technically infeasible. In previous work [1], we designed a set of depletion probes for human stool samples using a design strategy based solely on sequence abundance, completely agnostic of the microbiomal species present. Here, we show that the human-based probes are less effective when used with mouse cecal samples. However, adapting additional rRNA depletion probes specifically to cecal content provides both greater efficiency and consistency for MetaT analysis of mouse samples.