Hijacking of inflammasome responses by the complement system during Pseudomonas aeruginosa-Aspergillus fumigatus sur-infection
Hijacking of inflammasome responses by the complement system during Pseudomonas aeruginosa-Aspergillus fumigatus sur-infection
Khau, S.; Treps, L.; Ilango, G.; Riteau, N.; Couillin, I.; Togbe, D.; Bigot, J.; Balloy, V.; David, C.; Charrier Le Blan, M.; Fouquenet, D.; Vasseur, V.; Fontaine, T.; Pappworth, I.; Marchbank, K.; Paget, C.; Baranek, T.; Biquand, E.; Britto, C. J.; Guillot, L.; Si-Tahar, M.; Briard, B.
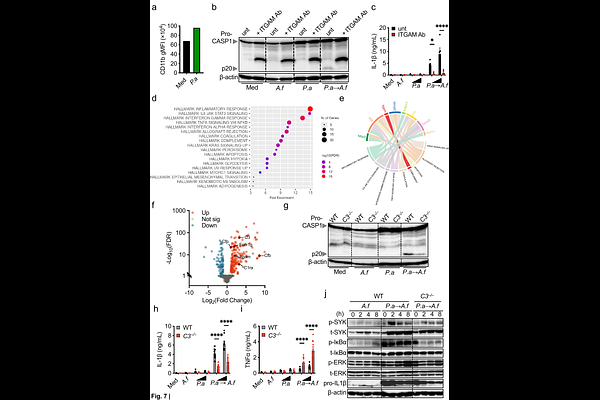
AbstractPatients with cystic fibrosis (pwCF) are highly susceptible to chronic pulmonary infections due to mutations in the CFTR gene. From early childhood, pwCF experience repeated lung infections and often develop chronic bacterial and/or fungal colonization. Among the most clinically relevant pathogens, Pseudomonas aeruginosa and Aspergillus fumigatus frequently co-infect and are associated with worse outcomes, including excessive IL-1{beta}-driven inflammation and accelerated lung function decline. Here we investigated the mechanisms underlying inflammasome overactivation during super-infection. We found that inflammasome hyperactivation occurred across macrophage populations, was independent of exogenous priming, and required live co-infection with both pathogens. P. aeruginosa and A. fumigatus cooperatively activated the NLRP3 inflammasome, and this response required both caspase-1 and caspase-8. Unexpectedly, gasdermin D was dispensable for IL-1{beta} release. Bacterial flagellin, type IV pili and the type III secretion system, as well as the fungal polysaccharide galactosaminogalactan (GAG), were each required for overactivation. Mechanistically, P. aeruginosa activated the MyD88-TLR pathway, enhancing macrophage responses and promoting ITGAM (CD11b) expression. Under fungal super-infection, macrophages secreted complement component C3, which may bound fungal surface and engaged the complement receptor C3R (CD11b/CD18). Downstream SYK and ERK signaling amplified inflammasome activation and IL-1{beta} release. Single-cell transcriptomic analysis of pwCF broncho-alveolar lavage and lung samples supported coordinated upregulation of complement and inflammasome pathways during bacterial-fungal infection. Together, these findings identify a complement-inflammasome signaling axis that drives pathological inflammation during bacterial-fungal co-infection in airways of pwCF and may represent a therapeutic target.