A bovine model of rhizomelic chondrodysplasia punctata caused by a deep intronic splicing mutation in the GNPAT gene


A bovine model of rhizomelic chondrodysplasia punctata caused by a deep intronic splicing mutation in the GNPAT gene
Boulling, A.; Corbeau, J.; Grohs, C.; Barbat, A.; Mortier, J.; Taussat, S.; Plassard, V.; Leclerc, H.; Fritz, S.; Leymarie, C.; Bourgeois-Brunel, L.; Ducos, A.; Guatteo, R.; Boichard, D.; Boussaha, M.; Capitan, A.
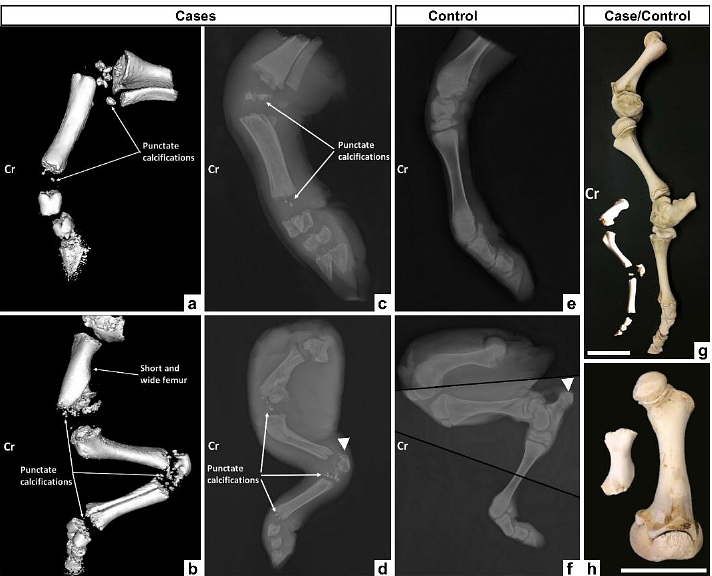
AbstractBackground Genetic defects that occur naturally in livestock species provide valuable models for investigating the molecular mechanisms underlying rare human diseases. Livestock breeds are subject to the regular emergence of recessive genetic defects, due to their low genetic variability, while their large population sizes provide easy access to case and control individuals, as well as massive amounts of pedigree, genomic and phenotypic information recorded for selection purposes. In this study, we investigated a lethal form of recessive chondrodysplasia observed in 21 stillborn calves of the Aubrac breed of beef cattle. Results Detailed clinical examinations revealed proximal limb shortening, epiphyseal calcific deposits and other clinical signs consistent with human rhizomelic chondrodysplasia punctata, a rare peroxisomal disorder caused by recessive mutations in one of five genes (AGPS, FAR1, GNPAT, PEX5 and PEX7). Using homozygosity mapping, whole genome sequencing of two affected individuals, and filtering for variants found in 1,867 control genomes, we reduced the list of candidate variants to a single deep intronic substitution in GNPAT (g.4,039,268G>A on Chromosome 28 of the ARS-UCD1.2 bovine genome assembly). For verification, we performed large-scale genotyping of this variant using a custom SNP array and found a perfect genotype-phenotype correlation in 21 cases and 26 of their parents, and a complete absence of homozygotes in 1,195 Aubrac controls. The g.4,039,268A allele segregated at a frequency of 2.6% in this population and was absent in 375,535 additional individuals from 17 breeds. Then, using in vivo and in vitro analyses, we demonstrated that the derived allele activates cryptic splice sites within intron 11 resulting in abnormal transcripts. Finally, by mining the wealth of records available in the French bovine database, we demonstrated that this deep intronic substitution was responsible not only for stillbirth but also for juvenile mortality in homozygotes and had a moderate but significant negative effect on muscle development in heterozygotes. Conclusions We report the first spontaneous large animal model of rhizomelic chondrodysplasia punctata and provide both a diagnostic test to counter-select this defect in cattle and interesting insights into the molecular consequences of complete or partial GNPAT insufficiency in mammals.