Influence of Sequencing Technology on Pangenome-level Analysis and Detection of Antimicrobial Resistance Genes in ESKAPE Pathogens
Influence of Sequencing Technology on Pangenome-level Analysis and Detection of Antimicrobial Resistance Genes in ESKAPE Pathogens
Frias-De-Diego, A.; Jara, M.; Lanzas, C.
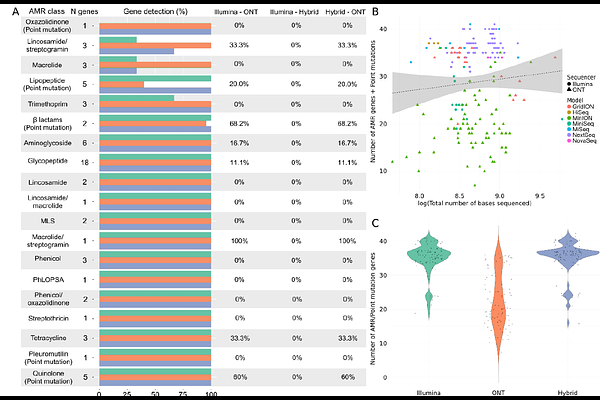
AbstractAs sequencing costs decrease, short-read and long-read technologies are indispensable tools for uncovering the genetic drivers behind bacterial pathogen resistance. This study explores the differences between the use of short-read (Illumina) and long-read (Oxford Nanopore Technologies, ONT) sequencing in detecting antimicrobial resistance (AMR) genes in ESKAPE pathogens (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter cloacae). Utilizing a dataset of 1,385 whole genome sequences and applying commonly used bioinformatic methods in bacterial genomics, we assessed the differences in genomic completeness, pangenome structure, and AMR gene and point mutation identification. Illumina presented higher genome completeness, while ONT identified a broader pangenome. Hybrid assembly outperformed both Illumina and ONT at identifying key AMR genetic determinants, presented results closer to Illumina\'s completeness, and revealed ONT-like pangenomic content. Notably, Illumina consistently detected more AMR-related point mutations than its counterparts. This highlights the importance of method selection based on research goals. Differences were also observed for specific gene classes and bacterial species, underscoring the need for a nuanced understanding of technology limitations. Overall, this study reveals the strengths and limitations of each approach, advocating for the use of Illumina for common AMR analysis; ONT for studying complex genomes and novel species, and hybrid assembly for a more comprehensive characterization, leveraging the benefits of both technologies.