Early mitophagy defects and impaired mitochondrial energy metabolism drive target organ damage progression: lessons from the Fabry heart
Early mitophagy defects and impaired mitochondrial energy metabolism drive target organ damage progression: lessons from the Fabry heart
GAMBARDELLA, J.; Fiordelisi, A.; Cerasuolo, F. A.; Buonaiuto, A.; Avvisato, R.; Viti, A.; Sommella, E.; Campiglia, P.; D'Argenio, V.; Prevete, N.; Pezone, A.; D'Apice, S.; Altobelli, G. G.; Varzideh, F.; Pande, S.; Paolillo, R.; Perrino, C.; Riccio, E.; Pisani, A.; Bianco, A.; Sadoshima, J.; Spinelli, L.; Santulli, G.; Sorriento, D.; Iaccarino, G.
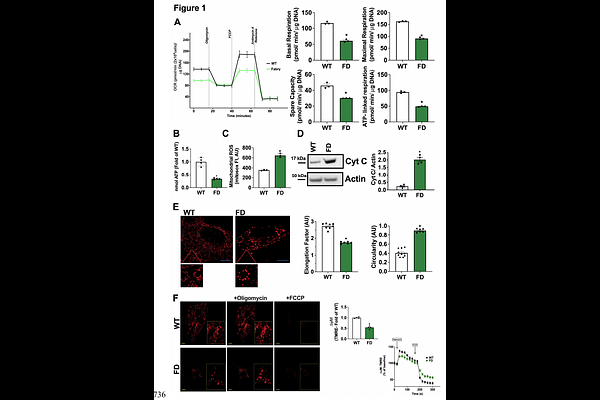
AbstractIncreased literature support the pathogenetic role of dysfunctional energetic metabolism in the setup and progression of organ damage and failure. Genetic diseases often offer the possibility to investigate pathogenetic mechanisms. In particular, excessive cardiac damage is the most frequent cause of mortality in Fabry disease (FD), a genetic condition caused by deficient -galactosidase A (GLA) activity, leading to globotriaosylceramide (Gb3) accumulation. Beyond Gb3 storage, metabolic alterations and mitochondrial dysfunction, supported by in vitro evidence or studies in other tissues, may contribute to FD cardiomyopathy. This study investigated, for the first time, the mechanisms of mitochondrial involvement in FD, its role in determining cardiac manifestations, and its potential as a therapeutic target. We used a humanized FD mouse model (R301Q-Tg/GLA knockout), along with derived embryonic fibroblasts and neonatal and adult cardiomyocytes, to assess mitochondrial function across the lifespan. FD cells showed impaired mitophagy, reduced mitochondrial respiration, and increased reactive oxygen species production. Importantly, this mitochondrial dysfunction exacerbated the lysosomal deficit in FD cells, forming a vicious cycle. In cardiomyocytes, these alterations progressed with age, leading to the accumulation of dysfunctional mitochondria, energetic failure, and, in adult hearts, terminal mitochondrial damage and apoptosis. These events ultimately result in cardiac remodeling and dysfunction, including hypertrophy and diastolic impairment. Indeed, L-arginine supplementation, which promotes NO/PGC-1-dependent mitochondrial rescue, prevented the development of cardiac abnormalities in FD mice. Our findings identify early mitochondrial dysfunction as a key driver of FD cardiomyopathy and support mitochondrial targeting, including L-arginine supplementation, as a promising adjuvant therapeutic strategy. The mechanistic link between lysosomal dysfunction, altered mitochondrial turnover, and energetic collapse emerges as a key targetable pathway in organ damage, extending beyond FD.