Hepatocyte Embryonic Ectoderm Development (Eed) Deficiency Causes Liver Injury, Fibrosis and Impacts Liver Regeneration
Hepatocyte Embryonic Ectoderm Development (Eed) Deficiency Causes Liver Injury, Fibrosis and Impacts Liver Regeneration
Ajouaou, Y.; Griffin, J.; Chen, C.; Chaffatt, S.; McManus, M.; Sadler, K. C.
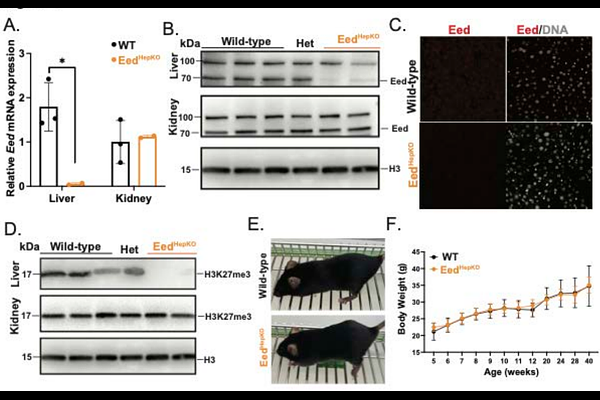
AbstractRegeneration depends on tightly coordinated transcriptional programs governed by a dynamic epigenetic landscape to regulate cell identity, proliferation, and tissue remodelling following injury. The liver is highly regenerative due to the ability to rapidly upregulate genes that drive the cell cycle and other genes important for regeneration. Trimethylation of histone 3 lysine 27 (H3K27me3) is deposited by the polycomb repressive complex 2 (PRC2) and many genes occupied by H3K27me3 in their promoters in uninjured livers become induced following PH. Here we test the hypothesis that depleting H3K27me3 by hepatocyte-specific deletion of Embryonic Ectoderm Development (EedHepKO), a key component of PRC2, changes the regenerative response in the liver. We show that Eed loss eliminates H3K27me3 in hepatocytes, resulting in reduced liver size, liver injury, increased hepatocyte death, proliferation and fibrosis associated with upregulation of cell cycle and fibrogenic genes. Though these mice are less likely to survive two-thirds partial hepatectomy than wildtype controls, those that do survive increase liver mass faster than WTs. Importantly, the genes that are occupied by H3K27me3 in control uninjured livers are upregulated in EEDHepKO and become further induced following PH. These data show that modulation of PRC2 activity removes repression of H3K27me3, patterning, induces liver injury, and alters regenerative outcomes, suggesting that precise control of PRC2 function could be harnessed to enhance regenerative capacity.