Identification of disease-specific alleles and gene duplications from 1,600 Haemophilus influenzae genomes using predicted protein analyses from an unsupervised language model and clinical metadata
Identification of disease-specific alleles and gene duplications from 1,600 Haemophilus influenzae genomes using predicted protein analyses from an unsupervised language model and clinical metadata
Palmer, P. R.; Earl, J. P.; Mell, J. C.; Koser, K. L.; Hammond, J.; Ehrlich, R. L.; Balashov, S. V.; Ahmed, A.; Lang, S.; Raible, K.; Wang, A. L.; Wigdahl, B.; Kaur, R.; Pichichero, M. E.; Dampier, W.; Ehrlich, G. D.
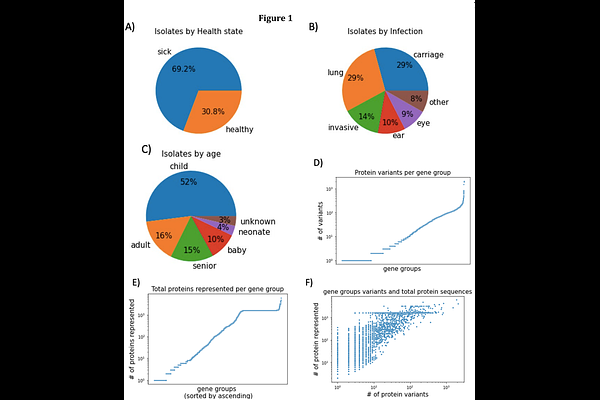
AbstractHaemophilus influenzae, a Gram-negative bacterium that is an obligate commensal of the human nasopharynx, is associated with both acute and chronic infections of the ears, adenoids, sinuses, and lungs, as well as pelvic inflammatory disease, sepsis, and meningitis. This diverse array of clinical disease phenotypes is due to H. influenzae's natural competence providing for frequent distributed gene exchange among strains during polyclonal colonizations. We performed whole genome sequencing (WGS) on approximately 1000 H. influenzae strains and combined these data with approximately 600 publicly available genomes. Clinical metadata including isolation site and disease were available for all strains. Our goal was to identify variations in protein sequences inferred from the WGS that correlate with the clinical metadata. We employed the alpha-fold unsupervised machine-learning model to all protein sequences. For each unique AA sequence, a numerical vector was generated reflecting the protein's biochemical properties. Multidimensional clustering was used to group variants into clusters by distance. For gene groups with two or more clusters, clinical metadata from the strain of origin was overlayed and subsequently tested for correlation with the clustering. This allowed identification of multiple gene groups with clusters of variants that correlated significantly with the patients' disease. COG category analysis showed that many of the significant gene groups were associated with antibiotic targets. The TbpA gene group type was found to be significantly correlated with disease type with three of five variant clusters having a 95 percent prevalence among strains that were isolated from the lungs of COPD patients or other lower pulmonary tract infections.