Diversity, functional classification and genotyping of SHV β-lactamases in Klebsiella pneumoniae
Diversity, functional classification and genotyping of SHV β-lactamases in Klebsiella pneumoniae
Tsang, K. K.; Lam, M. M. C.; Wick, R. R.; Wyres, K. L.; Bachman, M. A.; Baker, S.; Barry, K.; Brisse, S.; Campino, S.; Chiaverini, A.; Cirillo, D. M.; Clark, T. G.; Corander, J.; Corbella, M.; Cornacchia, A.; Cuenod, A.; D'Alterio, N.; Di Marco, F.; Donado-Godoy, P.; Egli, A.; Farzana, R.; Feil, E. J.; Fostervold, A.; Gorrie, C. L.; Gütlin, Y.; Hassan, B.; Hetland, M. A. K.; Hoa, L. N. M.; Hoi, L. T.; Howden, B.; Ikhimiukor, O. O.; Jenney, A. W.; Kaspersen, H.; Khokhar, F.; Leangapichart, T.; Ligowska-Marzeta, M.; Löhr, I. H.; Long, S. W.; Mathers, A. J.; McArthur, A. G.; Nagaraj, G.; Oaikhe
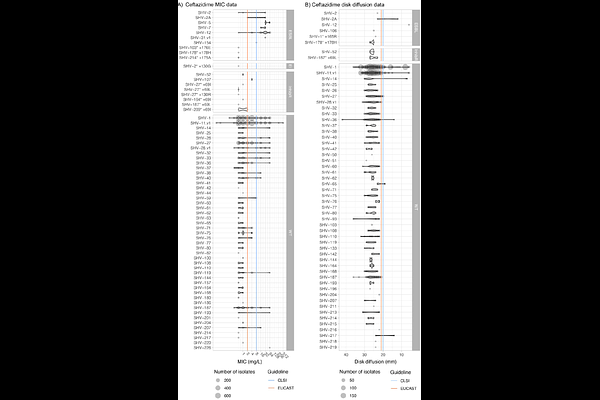
AbstractInterpreting phenotypes of blaSHV alleles in Klebsiella pneumoniae genomes is complex. While all strains are expected to carry a chromosomal copy conferring resistance to ampicillin, they may also carry mutations in chromosomal blaSHV alleles or additional plasmid-borne blaSHV alleles that have extended-spectrum {beta}-lactamase (ESBL) activity and/or {beta}-lactamase inhibitor (BLI) resistance activity. In addition, the role of individual mutations/amino acid changes is not completely documented or understood. This has led to confusion in the literature and in antimicrobial resistance (AMR) gene databases (e.g., NCBI\'s Reference Gene Catalog and the {beta}-lactamase database (BLDB)) over the specific functionality of individual SHV protein variants. Therefore, identification of ESBL-producing strains from K. pneumoniae genome data is complicated. Here, we reviewed the experimental evidence for the expansion of SHV enzyme function associated with specific amino-acid substitutions. We then systematically assigned SHV alleles to functional classes (wildtype, ESBL, BLI-resistant) based on the presence of these mutations. This resulted in the re-classification of 37 SHV alleles compared with current assignments in NCBI\'s Reference Gene Catalog and/or BLDB (21 to wildtype, 12 to ESBL, 4 to BLI-resistant). Phylogenetic and comparative genomic analyses support that; i) SHV-1 (encoded by blaSHV-1) is the ancestral chromosomal variant; ii) ESBL and BLI-resistant variants have evolved multiple times through parallel substitution mutations; iii) ESBL variants are mostly mobilised to plasmids; iv) BLI-resistant variants mostly result from mutations in chromosomal blaSHV. We used matched genome-phenotype data from the KlebNET-GSP Genotype-Phenotype Group to identify 3,999 K. pneumoniae isolates carrying one or more blaSHV alleles but no other acquired {beta}-lactamases, with which we assessed genotype-phenotype relationships for blaSHV. This collection includes human, animal, and environmental isolates collected between 2001 to 2021 from 24 countries across six continents. Our analysis supports that mutations at Ambler sites 238 and 179 confer ESBL activity, while most omega-loop substitutions do not. Our data also provide direct support for wildtype assignment of 67 protein variants, including eight that were noted in public databases as ESBL. We reclassified these eight variants as wildtype, because they lack ESBL-associated mutations, and our phenotype data support susceptibility to 3GCs (SHV-27, SHV-38, SHV-40, SHV-41, SHV-42, SHV-65, SHV-164, SHV-187). The approach and results outlined here have been implemented in Kleborate v2.4.1 (a software tool for genotyping K. pneumoniae from genome assemblies), whereby known and novel blaSHV alleles are classified based on causative mutations. Kleborate v2.4.1 was also updated to include ten novel protein variants from the KlebNET-GSP dataset and all alleles in public databases as of November 2023. This study demonstrates the power of sharing AMR phenotypes alongside genome data to improve understanding of resistance mechanisms.