Replicative senescence is ATM driven, reversible, and accelerated by hyperactivation of ATM at normoxia


Replicative senescence is ATM driven, reversible, and accelerated by hyperactivation of ATM at normoxia
Stuart, A. J.; de Lange, T.
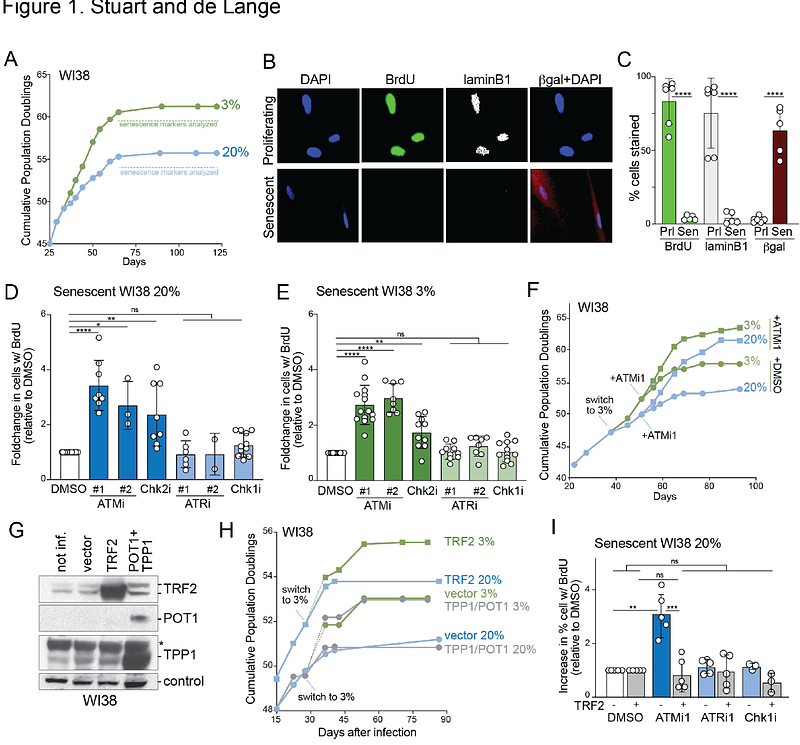
AbstractProgrammed telomere shortening limits tumorigenesis through the induction of replicative senescence. Here we address three long-standing questions concerning senescence. First, we show that the ATM kinase is solely responsible for the induction of replicative senescence. Senescence was delayed by ATM inhibition (ATMi) or overexpression of TRF2, the shelterin subunit dedicated to ATM repression. In contrast, there was no evidence for ATR signaling contributing to replicative senescence even when ATMi was combined with ATR inhibition. Second, we show ATMi can induce apparently normal cell divisions in a subset of senescent cells, indicating that senescence can be reversed. Third, we show that the extended replicative life span at low (physiological) oxygen is due to diminished ATM activity. At low oxygen, cells show a decreased ATM response to dysfunctional telomeres and genome-wide DSBs compared to 20% oxygen. As this effect could be reversed by NAC, the attenuated response of ATM to critically short telomeres and the resulting extended life span at low oxygen is likely due to ROS-induced formation of cysteine disulfide-bridges that crosslink ATM dimers into a form that is not activated by DSBs. These findings show how primary human cells detect shortened telomeres and reveal the molecular mechanism underlying the telomere tumor suppressor pathway.