Deep learning framework ChIANet predicts protein-mediated chromatin architecture across functional contexts
Deep learning framework ChIANet predicts protein-mediated chromatin architecture across functional contexts
Luo, H.; Wen, R.; Tang, L.; Chen, L.; Li, M.
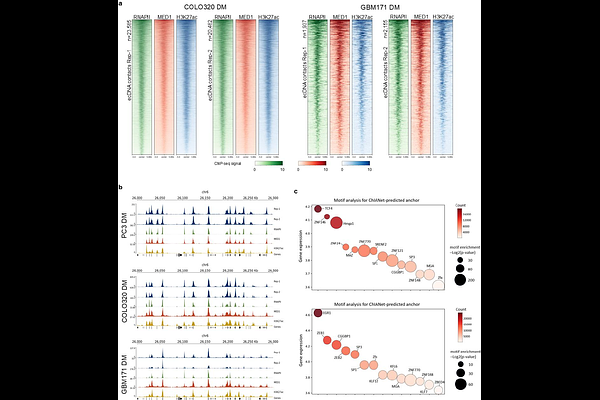
AbstractThe spatial organization of the genome is dynamically shaped by chromatin-binding proteins, yet how protein-mediated three-dimensional (3D) architectures are specified across functional contexts remains incompletely understood. Here we present ChIANet, a multimodal deep learning framework that enables de novo prediction of protein-mediated chromatin contact maps and loops from protein-binding profiles alone, using the reference genome sequence as a prior. By integrating transformer-based long-range modeling with multi-task learning, ChIANet accurately reconstructs protein-mediated 3D chromatin architectures and generalizes across diverse cellular contexts. Systematic application of ChIANet to CTCF, Cohesin and RNAPII across seven human cell types reveals that chromatin architectures follow conserved organizational principles while exhibiting pronounced context-dependent reconfiguration: CTCF- and Cohesin-mediated interactions predominantly support stable structural frameworks, whereas RNAPII-mediated loops display greater variability and are closely coupled to transcriptional programs and regulatory element activity. Functional analyses further uncover distinct regulatory biases and super-enhancer associations among the three proteins. Extending this framework to cancer genomes, ChIANet captures RNAPII-mediated chromatin looping networks associated with extrachromosomal DNA (ecDNA), revealing highly connected, transcription-associated architectures within amplified ecDNA regions across multiple cancer cell types. Together, these results demonstrate that protein-mediated 3D genome organization is not determined by protein identity alone but is flexibly shaped by functional context, regulatory targets and cellular environment, establishing ChIANet as a unified and scalable approach for decoding context-dependent principles of genome folding.