Neuronal endolysosomal acidification relies on interactions between transmembrane protein 184B (TMEM184B) and the vesicular proton pump
Neuronal endolysosomal acidification relies on interactions between transmembrane protein 184B (TMEM184B) and the vesicular proton pump
Wright, E. B.; Larsen, E. G.; Padilla-Rodriguez, M.; Langlais, P. R.; Bhattacharya, M. R. C.
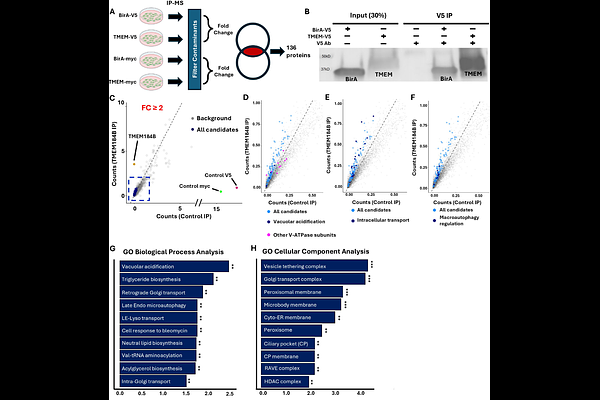
AbstractDisruption of endolysosomal acidification is a hallmark of several neurodevelopmental and neurodegenerative disorders. Impaired acidification causes accumulation of toxic protein aggregates and disrupts neuronal homeostasis, yet the molecular mechanisms regulating endolysosomal pH in neurons remain poorly understood. A critical regulator of lumenal acidification is the vacuolar ATPase (V- ATPase), a proton pump whose activity depends on dynamic assembly of its V0 and V1 subdomains. In this study, we identify transmembrane protein 184B (TMEM184B) as a novel regulator of endolysosomal acidification in neurons. TMEM184B is an evolutionarily conserved 7-pass transmembrane protein required for synaptic structure and function, and sequence variation in TMEM184B causes neurodevelopmental disorders, but the mechanism for this effect is unknown. We performed proteomic analysis of TMEM184B-interacting proteins and identified enrichment of components involved in endosomal trafficking and function, including the V-ATPase. TMEM184B localizes to early and late endosomes, further supporting a role in the endosomal system. Loss of TMEM184B results in significant reductions in endolysosomal acidification within cultured mouse cortical neurons. This alteration in pH is associated with impaired assembly of the V-ATPase V0 and V1 subcomplexes in the absence of TMEM184B, suggesting a mechanism by which TMEM184B promotes flux through the endosomal pathway. Overall, these findings identify a new contributor in maintaining endosomal function and provide a mechanistic basis for disrupted neuronal function in human TMEM184B-associated nervous system disorders.