Hidden Structural Bias in Proteomics: Sonication-induced Selective Fragmentation of Intrinsically Disordered Regions
Hidden Structural Bias in Proteomics: Sonication-induced Selective Fragmentation of Intrinsically Disordered Regions
Narita, M.; Yamakawa, T.; Nishimura, R.; Iwasaki, M.
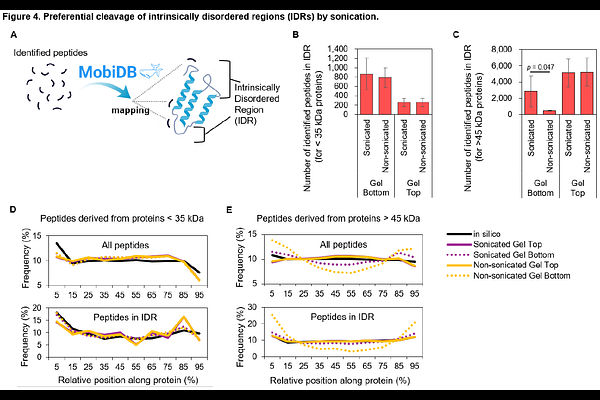
AbstractSonication is a fundamental technique in proteome sample preparation, primarily used for protein solubilization and shearing of genomic DNA. Although the mechanical shearing of DNA is well-characterized, its unintended impact on protein structural integrity remains a significant ''blind spot'' in high-throughput analytical workflows. In this study, we systematically investigated sonication-induced protein fragmentation by combining gel-based fractionation (PEPPI-MS) with sequence-level compositional analysis and bioinformatic mapping. Our results demonstrate that sonication does not significantly alter overall proteome identification or the recovery of membrane proteins; however, it induces extensive and non-random protein fragmentation. Sonication caused an approximately three-fold increase in the abundance of >45 kDa protein-derived fragments migrating into the <40 kDa fraction, and 1,620 high-molecular-weight (MW) proteins were uniquely detected in the lower-MW fraction upon sonication, an eight-fold increase over non-sonicated controls. Peptide-level amino acid composition analysis revealed subtle but directional shifts in the sonication-derived fragments. This residue-level signature is reinforced by two orthogonal structural analyses (MobiDB peptide-level mapping and protein-level profiling using metapredict V3 software), which show that sonication-susceptible proteins harbor more than twice the disordered content of length-matched controls (median 40% vs. 18%). This study identifies a previously unrecognized ''structural bias'' whereby intrinsically disordered region (IDR)-rich proteins are selectively compromised during sample preparation. Because these fragments are indistinguishable from enzymatic digestion products in conventional bottom-up proteomics, the underlying structural damage is effectively masked in global quantitative datasets, potentially distorting biological interpretations related to protein size, isoforms, and stability, particularly for IDR-rich classes, such as transcription factors and signaling molecules. We propose that optimizing and standardizing sonication parameters is essential for ensuring the accuracy and reproducibility of quantitative proteomic analyses.